使用GATK4获取SNP
1. 实验目的
GATK (Genome Analysis Toolkit),它是用于分析高通量测序数据的命令行工具集合,主要侧重于变体发现。本实验将使用使用GATK4获取SNP。
实验步骤为:准备参考基因组其他文件构成BWA索引,将样本ZW177与参考基因进行比对,对重复reads进行排序和标记生成.sorted.dedup.bam文件 ,IGV可视化BAM文件,在单个样本上运行GATK HaplotypeCaller,使用GenotypeGVCFs进行联合变异的获取,使用FreeBayes进行联合变异获取,过滤SNP确保只有高质量的变异数据被用于后续的分析,基本的统计和比较不同的集合等过程。
2. 实验准备
2.1 实验平台
Linux JSvr01 3.10.0-1160.80.1.el7.x86_64 #1 SMP Tue Nov 8 15:48:59 UTC 2022 x86_64 x86_64 x86_64 GNU/Linux
2.2 数据简述
| 数据 | 来源 |
|---|---|
 |
/work/cb-data/Variants_workshop_2020/*.fastq.gz |
| genome.fa | /work/cb-data/Variants_workshop_2020/genome |
 |
/work/cb-data/Variants_workshop_2020/premade_gvcf/ |
 |
/work/cb-data/Variants_workshop_2020/processed_bams/ |
 |
/work/cb-data/Variants_workshop_2020/scripts |
2.3 软件配置
| 软件 | 版本 | 来源 |
|---|---|---|
| bwa | 0.7.17-r1188 | Linux |
| GATK(自行设置路径) | 4.1.9.0 | Linux |
| Samtools | 1.7 | Linux |
| vcftools | 0.1.15 | Linux |
| FileZilla | 3.65.0 | https://filezilla-project.org/ |
| IGV | 2.6.3 | https://data.broadinstitute.org/igv/projects/downloads/2.6/ |
3. 实验内容
本实验步骤为:准备参考基因组其他文件构成BWA索引,将样本ZW177与参考基因进行比对,对重复reads进行排序和标记生成.sorted.dedup.bam文件 ,IGV可视化BAM文件,在单个样本上运行GATK HaplotypeCaller,使用GenotypeGVCFs进行联合变异的获取,使用FreeBayes进行联合变异获取,过滤SNP确保只有高质量的变异数据被用于后续的分析,基本的统计和比较不同的集合等过程。
3.1 Data used in the exercise
我们将使用 黑腹果蝇WGS配对末端Illumina数据与NCBI种质SRR1663608,SRR1663609,SRR1663610,SRR1663611,分别对应于样品ZW155,ZW177,ZW184和ZW185。为了加快计算速度,最初每个样本约1000万个读取对的数据已被下采样50%。
该练习包括这几个步骤:从 WGS Illumina 读取到 GATK 最佳实践中指定的四样本变体获取。要获取所有四个样本中的变体,需要对四个 FASTQ 文件对中的每一个执行从读取对齐到单倍型获取的所有步骤。在具有大内存的多 CPU 计算机上,可以并行启动此类运行。
3.2 Log in to your workshop machine
3.3 General tips
检查所有提供的 shell 脚本,例如,在文本编辑器(如nano或vim)中打开它们。阅读解释性评论。注意使用环境变量来简化和泛化脚本。例如,下面定义一个变量ACC:
1 | |
每当脚本中出现 $ACC 或 ${ACC} 时,都会被解释为 SRR1663609 。请注意将长行分成以“”字符结尾的较小部分的技术。对于bash来说,这仍然是一条很长的线,但对我们来说更容易阅读。
使用 top 命令监控活动的进度,最好在单独的窗口中运行:
1 | |
这将显示动态更新的流程列表,最活跃的进程位于顶部。由于GATK工具是用Java编写的,因此 将看到最多的过程将是称为java的Java虚拟机。 在对齐阶段,要寻找的过程将是称为 bwa 的 BWA 对准器。 顶部列表中没有任何活动进程(消耗 CPU 时间)将表示 正在运行的任何脚本完成(或崩溃)。注意不同运行的内存使用情况(%MEM 列)。
查看日志文件。每次运行脚本时,所有命令的屏幕输出都会保存到日志文件(例如,脚本 .log )。尽管写入该文件的消息有时听起来很神秘,但它们通常允许用户确定当前正在运行计算的哪个阶段。它还包含有用的计时信息(各个阶段的开始和结束日期,经过的时间,预计到达时间)。若要查看日志文件,可以使用以下命令之一:
1 | |
当然, 也可以通过在文本编辑器中打开它来查看整个文件。
查看工作目录 (/$USER)
随着运行的进行,正在生成各种中间文件。偶尔执行 ls -al
将允许
查看这些文件以及它们的大小如何增加。如果即使脚本似乎已结束,也看不到预期的输出文件,通常意味着出了问题。检查屏幕日志文件以查找错误消息。
可以断开连接。如果一个步骤花费的时间超过愿意等待的时间,可以断开与 VNC 或screen会话的连接(在 VNC 中,单击 VNC 窗口右上角的叉号,但不要 “注销”!所有程序和窗口将继续运行, 可以在以后重新连接时检查结果。此外,如果 通过 ssh 客户端(而不是 VNC)工作并且没有screen,只要 正在运行的脚本是通过 nohup 在后台提交的, 就可以安全地注销 ssh 会话(按照本练习中的建议)。下次登录到计算机时,脚本仍将运行(或已完成)。
3.4 Fetch input files and scripts to your local scratch directory
准备所需的文件,放至工作目录下。
(1) 创建文件夹
1 | |
(2) 复制文件至工作目录
1 | |
需要注意的是,脚本文件需要可执行权限,可在/scripts目录下使用chmod u+x *.sh加权限。
3.5 Prepare reference genome
子目录基因组中的文件 genome.fa 是我们将读取对齐的参考。在启动管道之前,需要为BWA对准器索引基因组。此外,需要总结所有染色体的长度信息。
(1) 修改脚本权限
1 | |
(2) 修改脚本路径
1 | |
(3) 运行脚本
1 | |
该脚本将在后台运行,大约需要 10 分钟才能完成(在单独的窗口中使用 top -u $USER 来监视进程)。
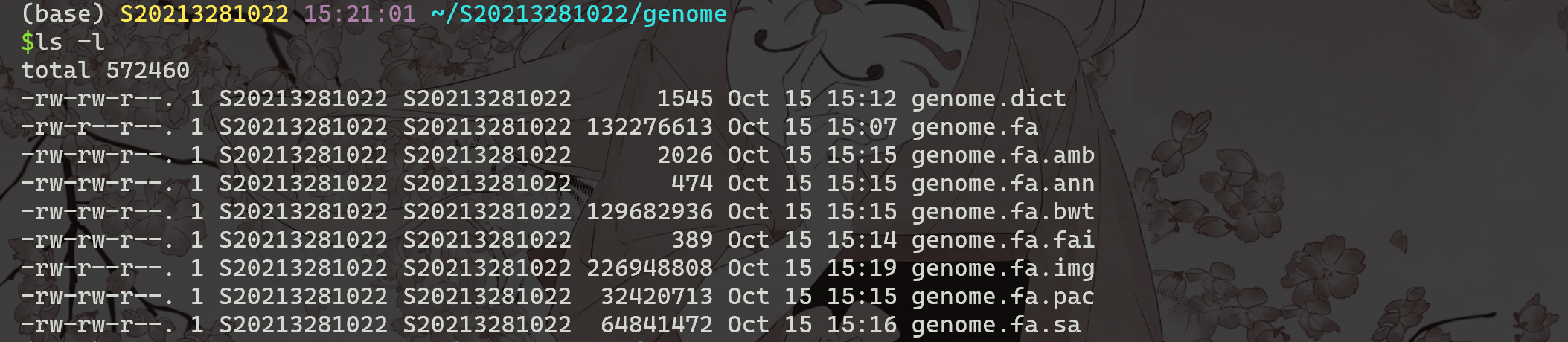
完成后,列出子目录基因组的内容 。文件genome.fa.fai 和 genome.fa.dict 是简单的文本文件,总结了原始FASTA文件中的染色体大小和起始字节位置。这与其他文件构成 BWA 索引。
(4) 查看工作日志
1 | |
(5) 查看提供随机访问fasta/fastq文件接口的.fai文件
1 | |
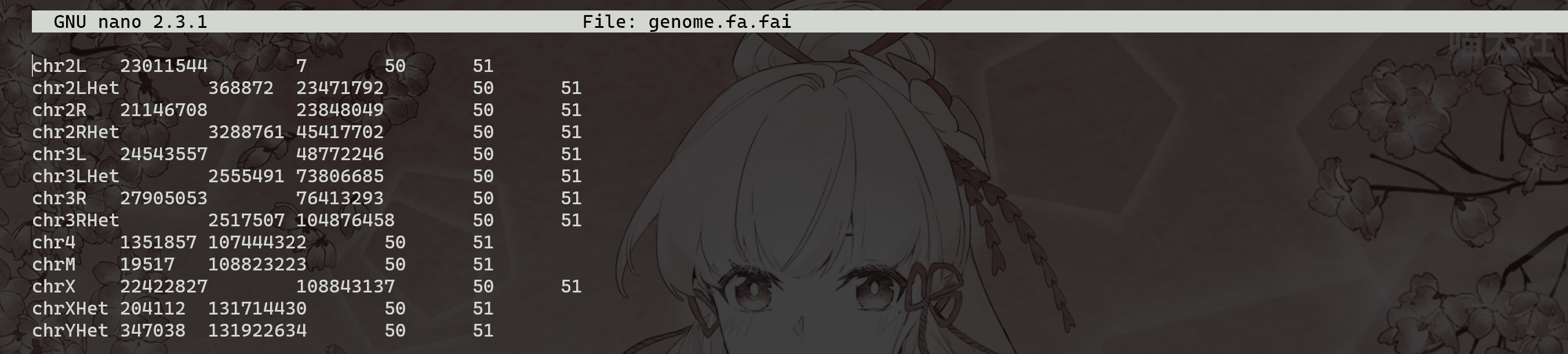
fai的格式:以,fasta 5列,fastq 6列:
- NAME Name of this reference sequence
- LENGTH Total length of this reference sequence, in bases
- OFFSET Offset in the FASTA/FASTQ file of this sequence’s first base
- LINEBASES The number of bases on each line
- LINEWIDTH The number of bytes in each line, including the newline
- QUALOFFSET Offset of sequence’s first quality within the FASTQ file
(6) 查看用来reference genome序列的dict文件
1 | |
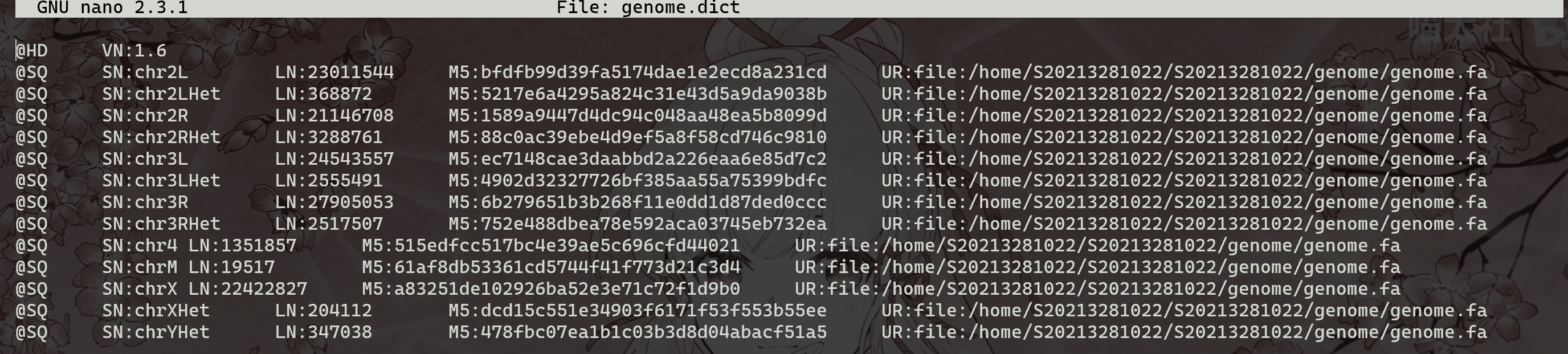
@SQ用于分割每一条染色体信息,第一列是染色体信息,第二列是染色体名字,第三列是M5码,第四列是来源 。
3.6 Align reads to reference
使用脚本aln_bwa.sh,将SRR1663609(样本ZW177)与参考基因进行比对,它立即将其转换为 BAM 格式的压缩二进制文件,相当于 SAM,但小了几倍。由于这种管道机制,bwa mem 的大型中间输出文件不需要写入磁盘。
在我们的练习中,每个样本在一个 Illumina 泳道上测序一次。
(1) 修改脚本权限
1 | |
(2) 执行脚本
1 | |
(3) 查看SRR1663609.bam
1 | |

3.7 Sort and mark duplicates
在此步骤中,在先前获得的BAM文件中发现的任何重复片段现在将被标记为在下游分析中忽略,并且将根据基因组坐标对比对进行排序。生成的排序 BAM 文件也将编制索引。所有这些操作现在都由一个名为 MarkDuplicatesSpark 的单个 GATK 工具执行。
运行的结果将是文件 SRR1663609.sorted.dedup.bam ,并附有索引文件 SRR1663609.sorted.dedup.bai 。
(1) 修改脚本权限
1 | |
(2) 修改脚本
1 | |
1 | |
(3) 执行脚本
1 | |
脚本运行近5分钟。

(4)查看目录中文件
1 | |
生成了.sorted.dedup.bam文件,以及索引文件.sorted.dedup.bai文件
(5) 使用samtools命令查看获取到的文件的比对状态汇总
检查获取的文件的对齐统计信息摘要,运行 samtools 命令。
1 | |

1 | |
3.8 Visualize the alignments using IGV
(1) 利用FilleZilla下载文件
SRR1663609.sorted.dedup.bam,SRR1663609.sorted.dedup.bam.bai,genome.fa
(2) 使用IGV可视化SRR1663609.sorted.dedup.bam
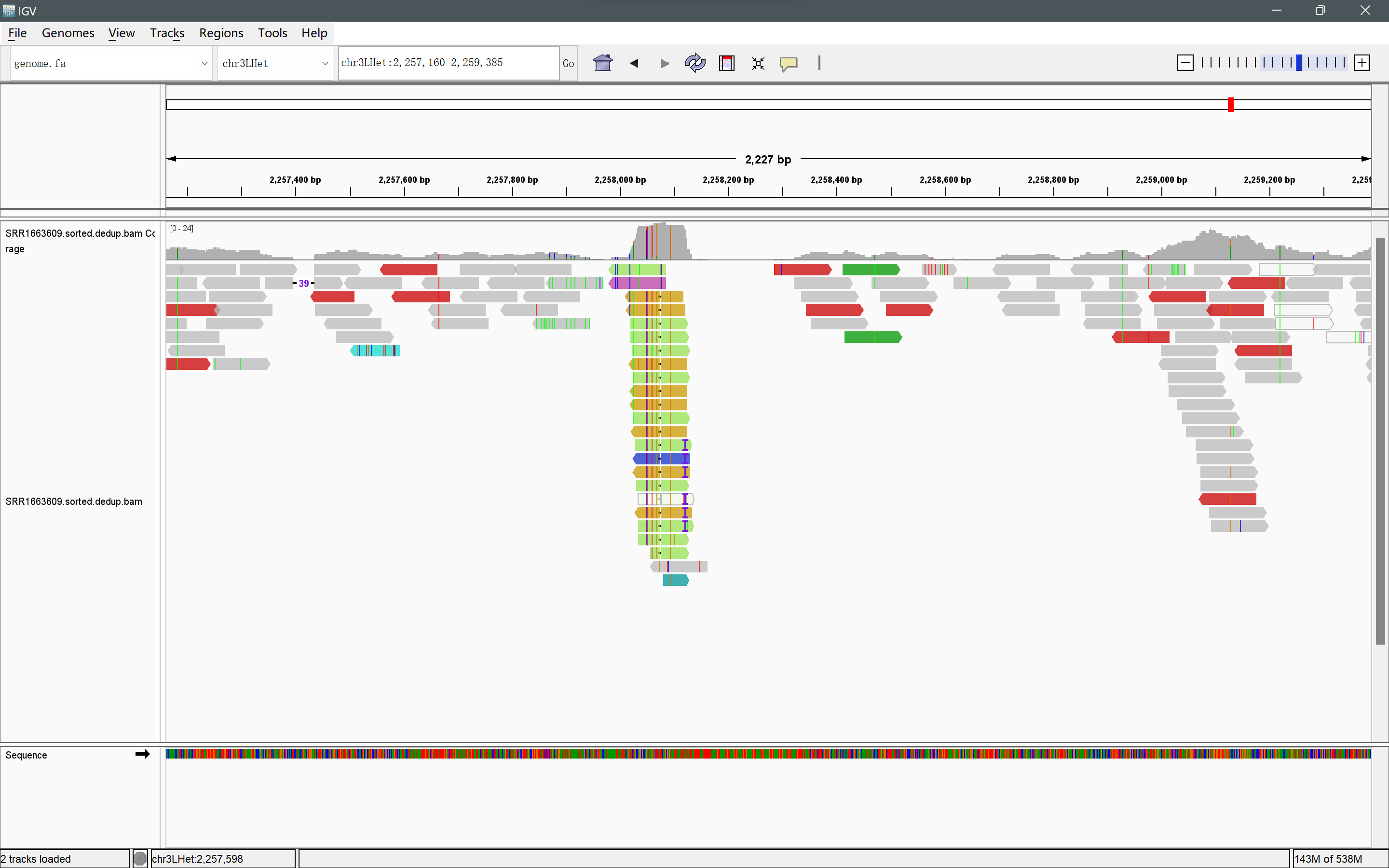
在IGV图中,灰色表示insert size正常,是常规reads。红色表示insert size大于预期,表示可能存在缺失突变。蓝色表示insert size小于预期,表示可能存在插入突变。紫色的部分说明了存在插入碱基,数字表示插入碱基的个数。当覆盖轨道为彩色时,表示核苷酸与参照碱基比对的差异查过20%,可以根据这个,判断这些点位的SNP。
本图出现较多的paired end reads中成对两条reads比对到了不同的染色体Mapping在chr3LHet:2,258,070bp左右,并且chr3LHet:2,257,400bp左右有一个read存在39bp的碱基插入。
3.9 Run GATK HaplotypeCaller on individual samples
在此步骤中,我们将在BAM文件上运行HaplotypeCaller,以生成感兴趣基因组区域中每个位点的每个样本的基因型似然信息。通常,这个区域将是整个基因组。为了节省时间,我们将专注于一条染色体chr2R。预期结果将是文件SRR1663609.g.vcf ,其中包含该样本的GVCF格式的中间基因分型数据 。
(1) 修改脚本权限
1 | |
(2) 修改脚本
1 | |
1 | |
(3)运行脚本
1 | |

此步骤的估计运行时间为 1 小时,并且必须对其他 3 个样本重复此操作。出于本练习的目的,我们建议至少一个示例使用 hc.sh 和脚本运行此步骤,并将预制的 .g.vcf 文件和相应的索引文件 .g.vcf.idx 复制到目录中。
(4) 查看 .g.vcf文件
1 | |
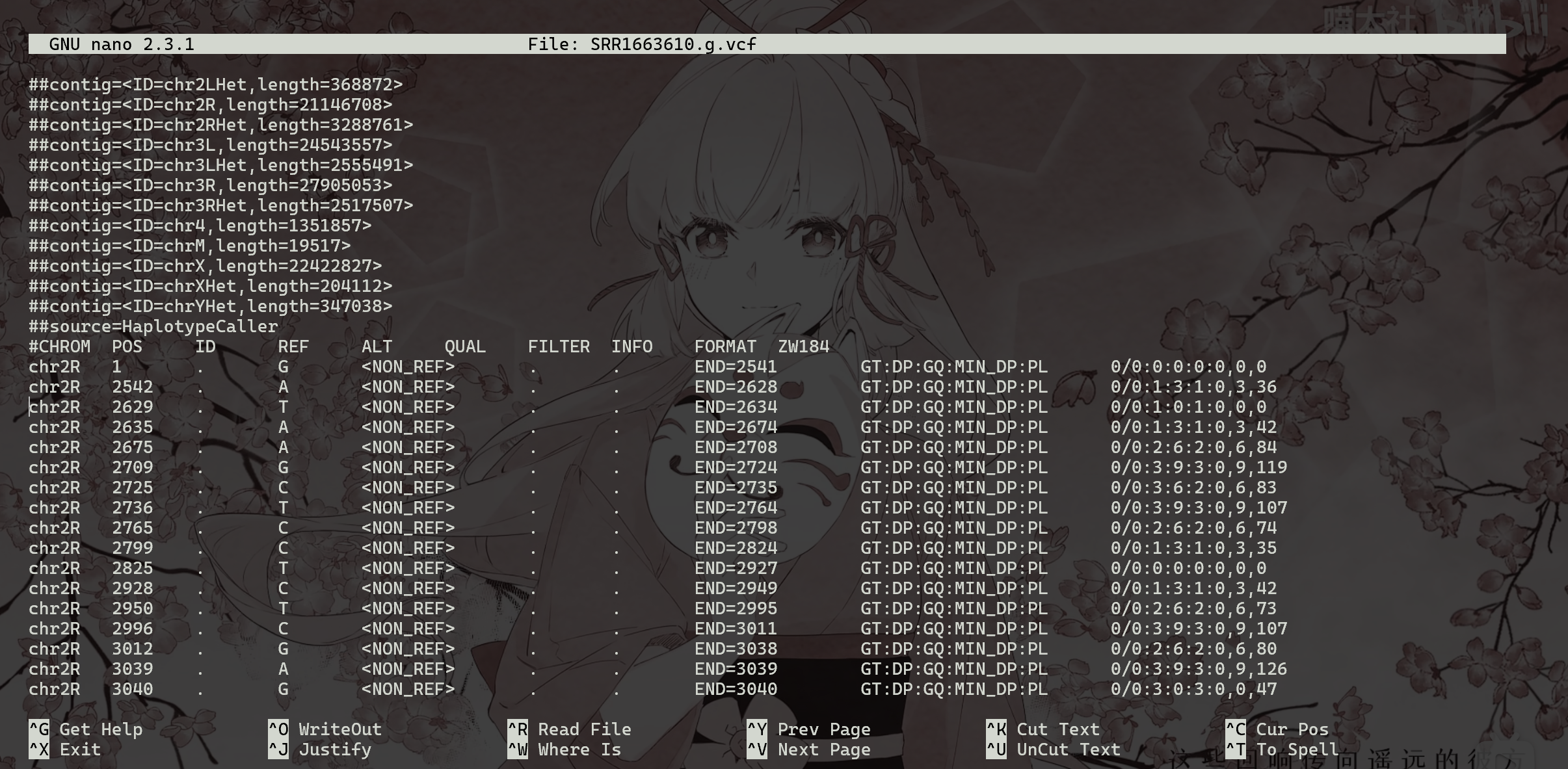
VCF格式用于记录变异位点(SNP/InDel)的文件格式。
#开头注释部分
无#开头 主体部分,主体部分包含10列数据,主体部分每一行代表一个变异位点信息。
主体部分10列代表的意义:
1、CHROM 参考序列名 2、POS 变异位点所在的left-most位置(1-base position)。(发生变异的的位置的第一个碱基所在) 3、ID 变异位点ID。同时对应着dbSNP数据库中的ID,若没有,使用默认使用 。 4、REF 参考序列的等位基因(Allele)(等位碱基,即参考序列该位置的碱基类型及碱基数量) 5、ALT 变异位点的等位基因,若有多个,则使用逗号分隔。(变异位点碱基) 6、QUAL 变异位点的质量。Phred格式的数值,代表着此位点是纯合的概率。此值越大,概率越低,代表 着此位点是变异位点的可能性越大。 7、FILTER 此位点是否要被过滤掉。如果是PASS,则表示此位点可以考虑为变异位点。 8、INFO 变异位点的相关信息。 9、FORMAT 变异位点的格式,如GT:AD:DP:GQ:PL。10、SAMPLEs各个样本的值,由BAM文件中@RG下的SM标签所决定。这些值对应着第九列的各种格式。 不同格式的值用冒号分开。每个样本对应着一列;多个样本则对应着多列,这种情况下列的数量 会超过10列。
(5) 查看目录文件
1 | |
3.10 Joint variant calling with GenotypeGVCFs
在中间样本级文件 *.g.vcf 结果可用于联合获取所有四个样本的变体之前,必须 使用 GATK4 的工具 CombineGVCFs 将它们合并为单个多样本 g.vcf 文件。
(1) 修改脚本权限
1 | |
(2) 修改脚本
1 | |
(3) 运行合并脚本
1 | |
运行时间:5-6 分钟。生成all.vcf文件 。

1 | |
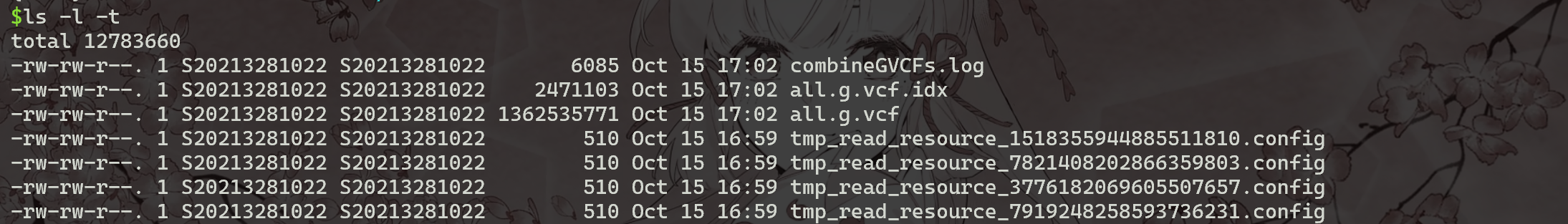
(4) 修改联合SNP的获取的脚本
1 | |
1 | |
(5) 获取联合变异
1 | |
运行时间:5-6 分钟。生成all.vcf文件 。

(6) 查看all.vcf文件
1 | |
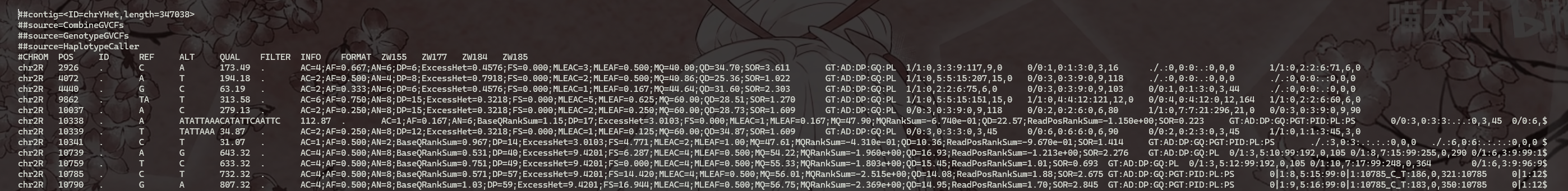
3.11 Joint variant calling using FreeBayes
与GATK的HaplotypeCaller类似,FreeBayes使用基于单倍型的方法来检测变体,尽管实现方式不同。FreeBayes 的输入由所有涉及的样本的对齐 BAM 文件组成。我们将使用在本练习前面获得的已处理的 BAM 文件。结果将是变体文件 fb.vcf 。
(1) 修改脚本权限
1 | |
(2) 修改脚本
1 | |
(3) 运行脚本
1 | |
(4) 检查生成的fb.vcf文件
1 | |

检查生成的 VCF 文件时,注意 FreeBayes 生成的注释字段中的参数通常与 GATK 获取方发出的参数不同。
3.12 Filter variants
在前面的步骤中获得的 VCF 文件是原始结果,可能包含大量误报,具体取决于获取中使用的严格选项。由于获取步骤非常耗时,因此通常建议将这些选项设置为发出一组包含的变体,然后根据各种参数过滤此组。在GATK,选项 –stand-call-conf 控制要输出的变体的质量下限阈值(VCF 的 QUAL 字段)。此选项应设置为某个低值。
(1) 从原始 all.vcf中提取QUAL大于60的变体子集
可以使用许多不同的工具完成原始变体集的过滤。在很多情况下,人们可以简单地使用标准的Linux文本解析工具,如grep,awk或sed。例如,要从原始 all 中提取 QUAL 大于 60 的变体子集.vcf我们可以使用以下命令:
1 | |
1 | |
(2) 使用VariantFiltration过滤
GATK提供了一个名为VariantFiltration的工具,它允许更复杂的过滤模式。使用此工具的示例脚本称为 filter_vcf.sh 。检查此脚本时,您会注意到应用于 SNP 的过滤条件与应用于插入缺失的过滤条件不同。为此,首先将 SNP 和插入缺失提取到单独的文件中,对这些文件进行过滤,然后将 SNP 和插入缺失过滤的文件合并回单个过滤文件。
1 | |
1 | |
1 | |

运行时间: 3 分钟。
1 | |
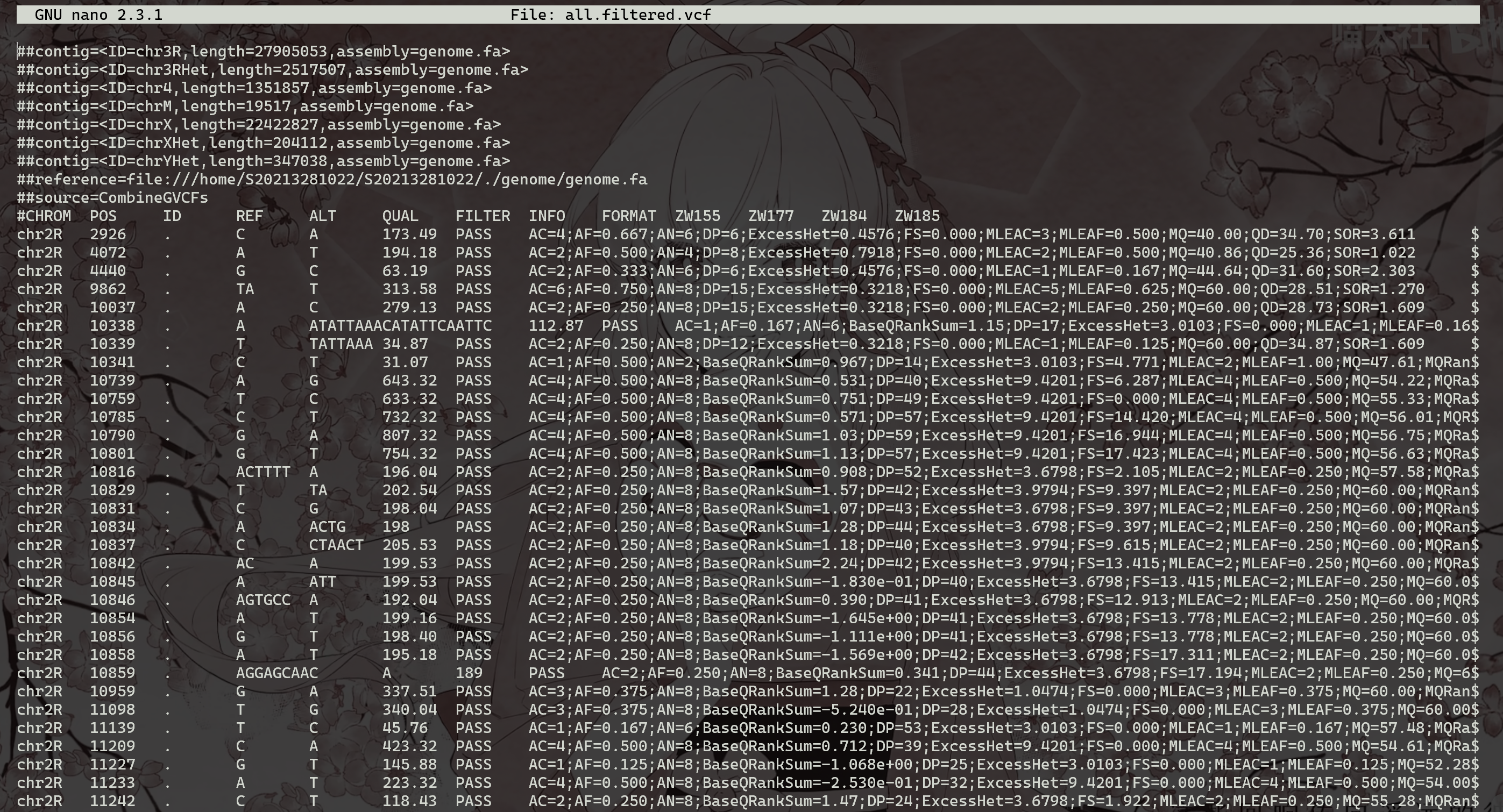
过滤后的VCF文件将称为 all.filtered.vcf (相应的索引文件*.vcf.idx 也将被创建)。其他中间文件(具有单独的 SNP、插入缺失、过滤连同它们的索引)也会产生——这些可能会被删除。检查已过滤的VCF文件。请注意“筛选器”字段中的更改。而不是点“.”(无过滤信息),此字段现在将包含标志 PASS (通过过滤器的变体)和my_snp_filter 或 my_indel_filter (这两个字符串都是在过滤命令中定义的)–标记未通过相应过滤器的变体。
3.13 Basic stats and comparison of variant sets
(1) 使用Linux命令
给定一个VCF文件,其最简单的属性可以通过运行标准的Linux文本解析工具获得。例如,若要获取文件中的变体数,请运行以下命令:
1 | |
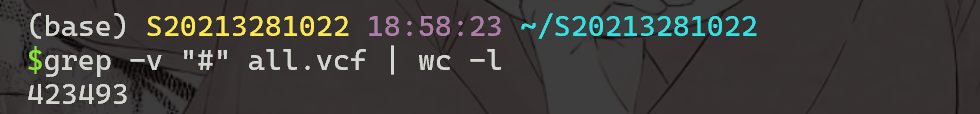
grep 过滤掉标题行并将其输出通过管道传输到 wc -l 中,后者计算剩余的行并在屏幕上显示结果)。要提取位于染色体chr2R上位置10000和20000之间的位点并将其保存在文件中,只需运行:
1 | |
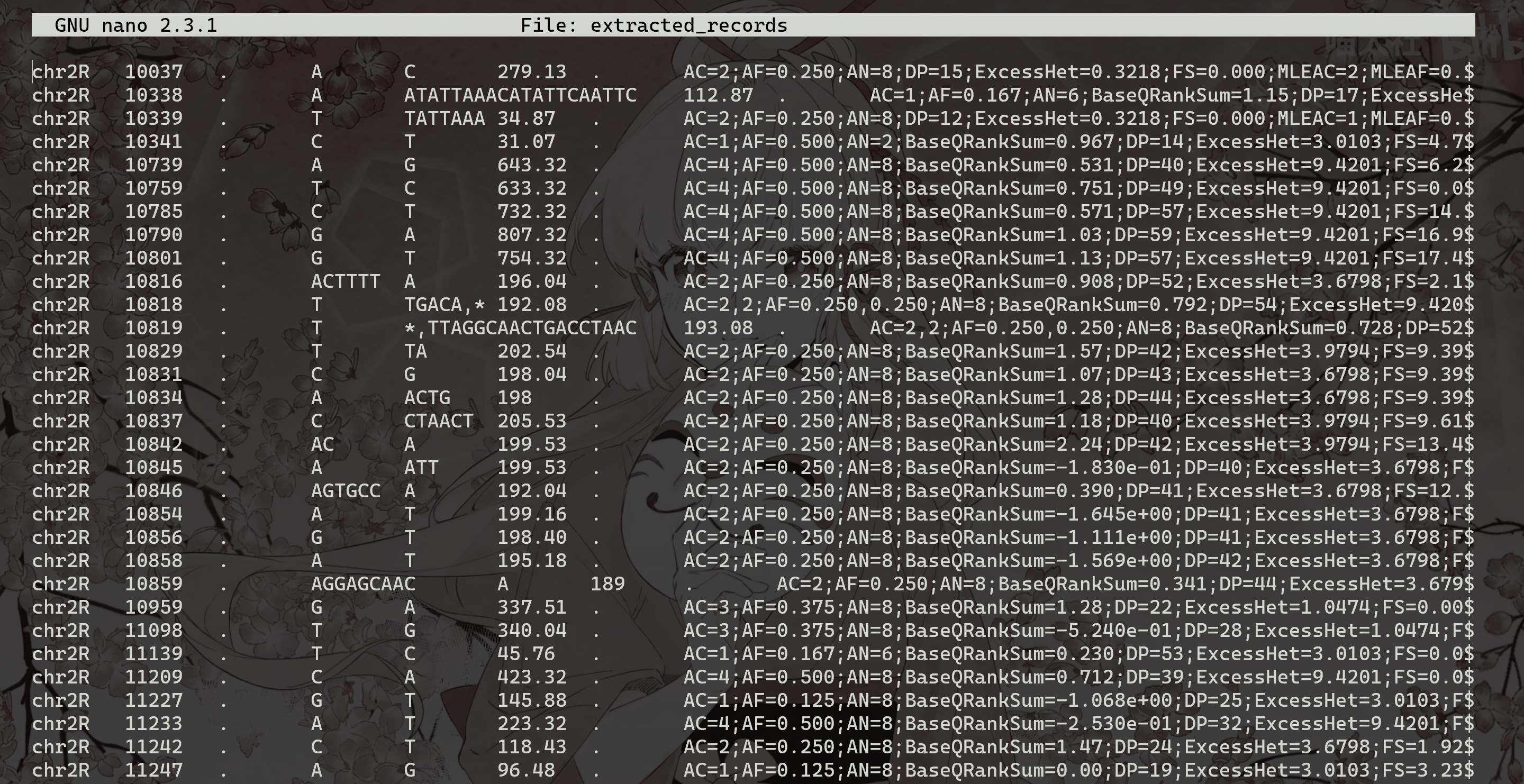
(请注意,在这种情况下,染色体条件$1==“chr2R”并不是真正需要的,因为我们的VCF文件只包含chr2R的数据,但是,需要更一般的输入)。要快速找出有多少变体通过了过滤,只需键入:
1 | |

存在417825变体。
(2) 使用GATK4的 Concordance 和 GenotypeConcordance 工具
GATK4 提供了有趣的函数,一致性和基因型一致性,以汇总变体集的各种统计数据,并将其与从相同数据获得的另一个变体集进行比较,但使用不同的方法,例如。基于此功能的脚本var_compar.sh将比较任意两个VCF文件,例如all.vcf和fb.vcf(分别使用GATK4和FreeBayes获得):
1 | |
1 | |
1 | |

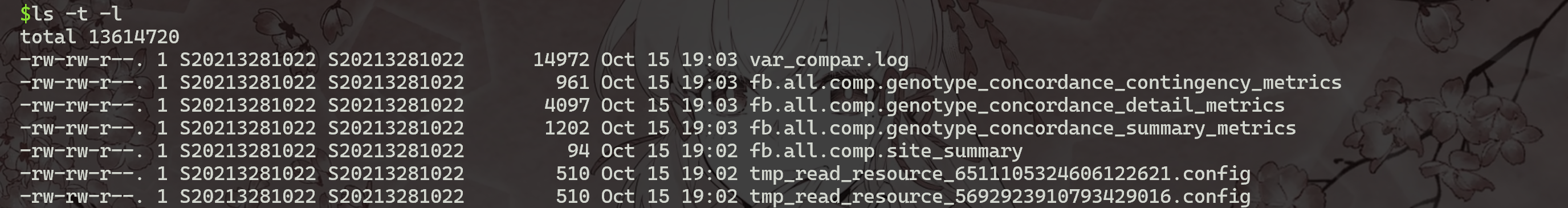
1 | |
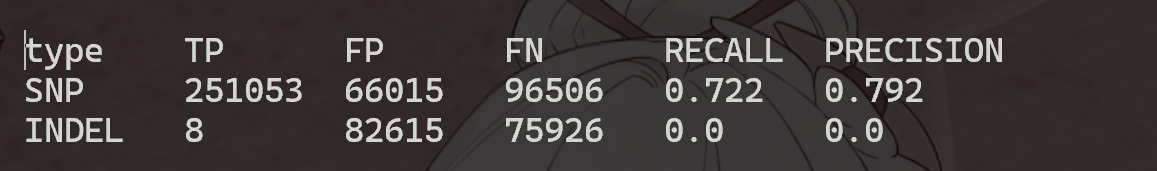
从图中可知,使用GATK4,以all.vcf为原假设,在InDel中,大部分都是假阳性,只有8个是真阳性。 而使用FreeBayes,以fb.vcf为原假设,其中有79.2%的SNP都是真阳性,少部分为假阳性。
1 | |
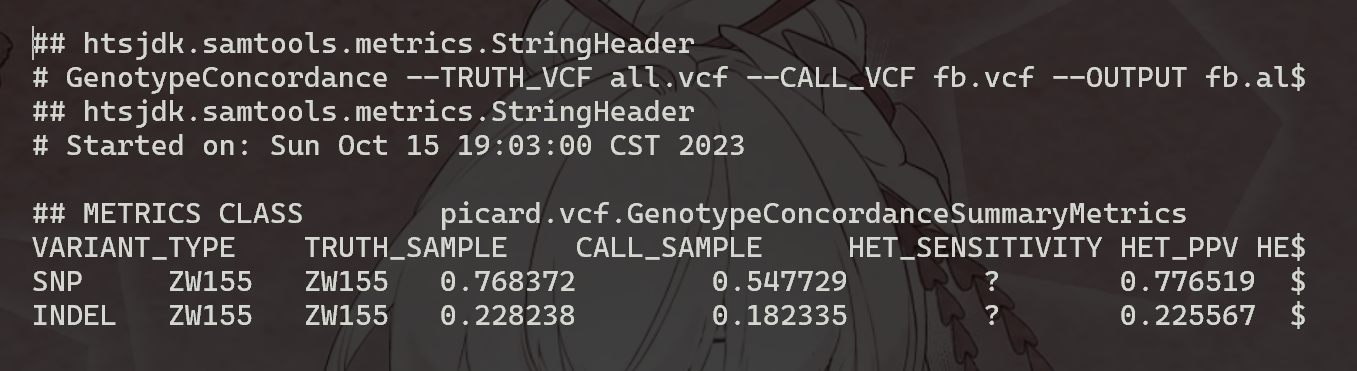
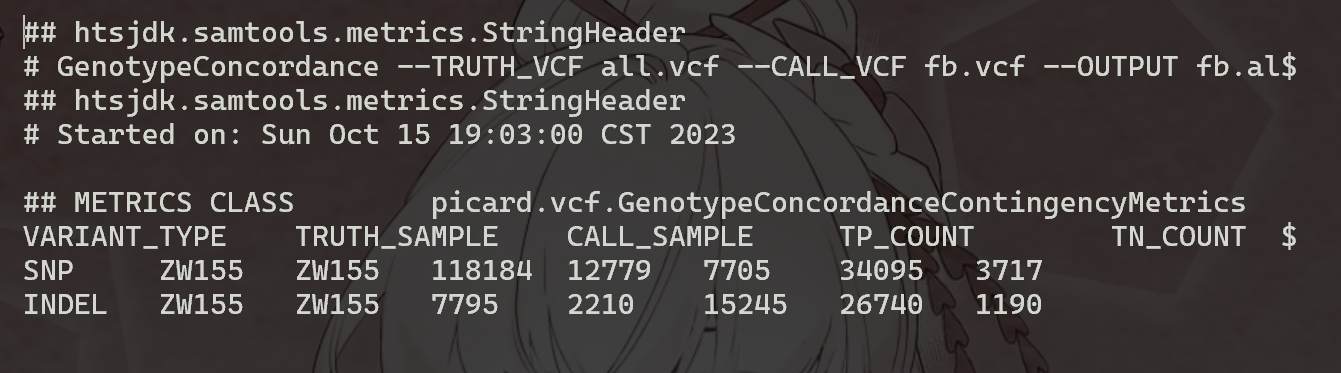
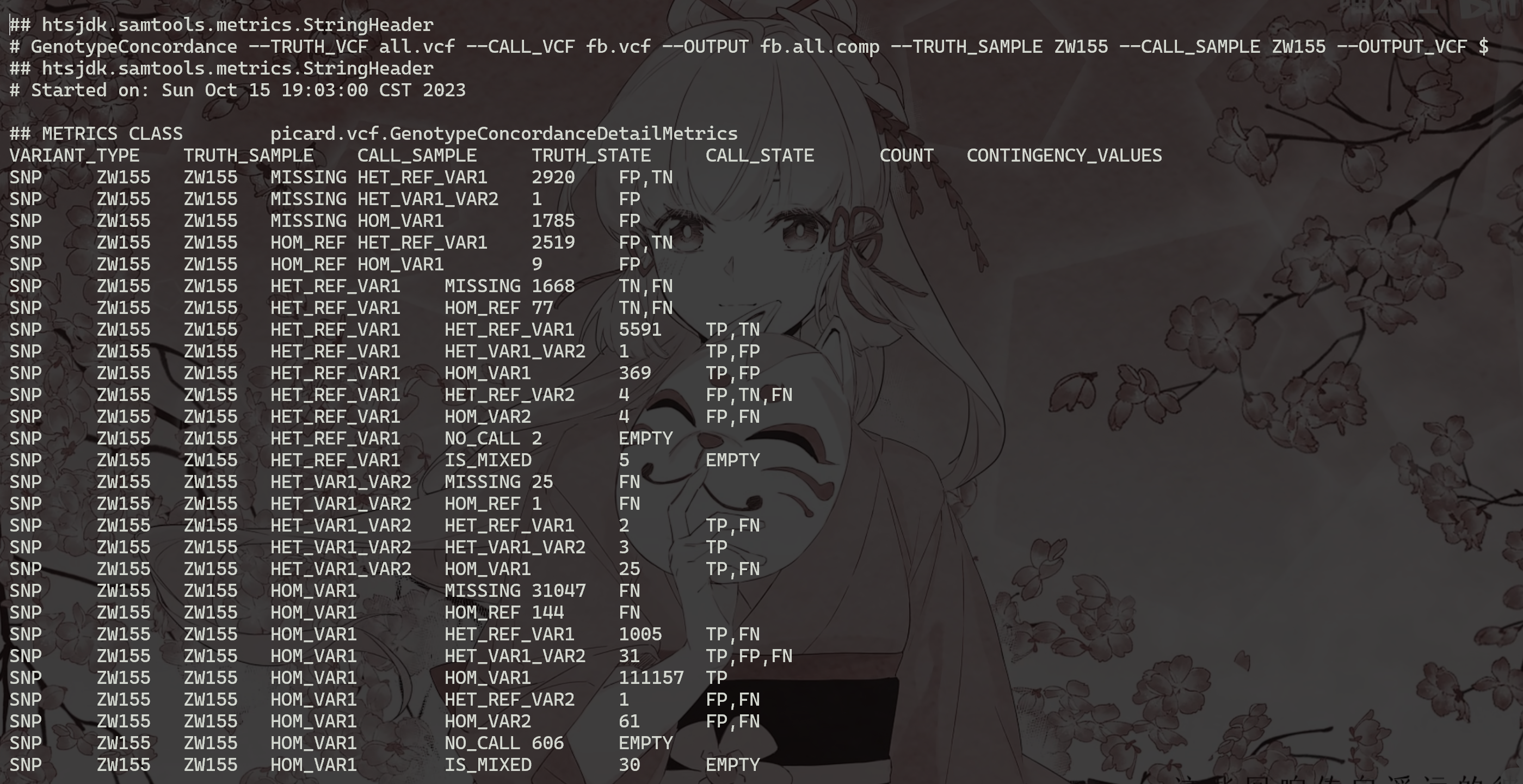
上面的命令将生成四个文件fb.all.comp.site_summary,其中包含站点一致性摘要,以及另外三个文件 (fb.all.comp.genotype_concordance_summary_metrics,fb.all.comp.genotype_concordance_detail_metrics,fb.all.comp.genotype_concordance_contingency_metrics),并提供更详细的分析ZW155基因型一致性。
(3) 使用 vcftools
vcftools 是用于分析和操作VCF文件的流行工具包。以下是一些使用示例(在VCF文件上尝试):
1 | |
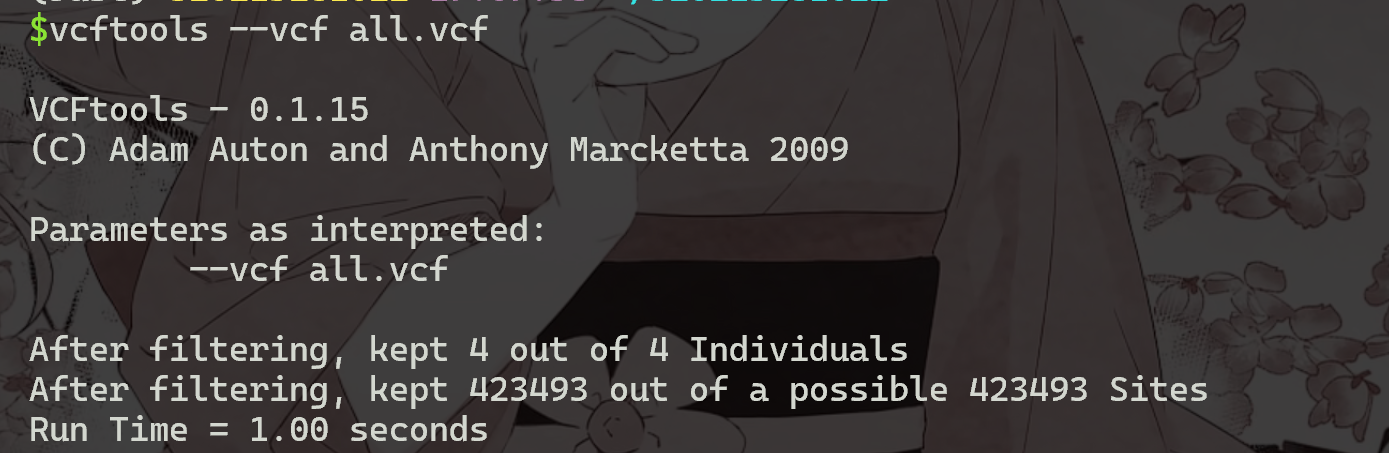
显示有四个样本,共423493个SNP。
提取变体子集(chr2R,1M和2M之间)并将其写入新的VCF文件 :
1 | |
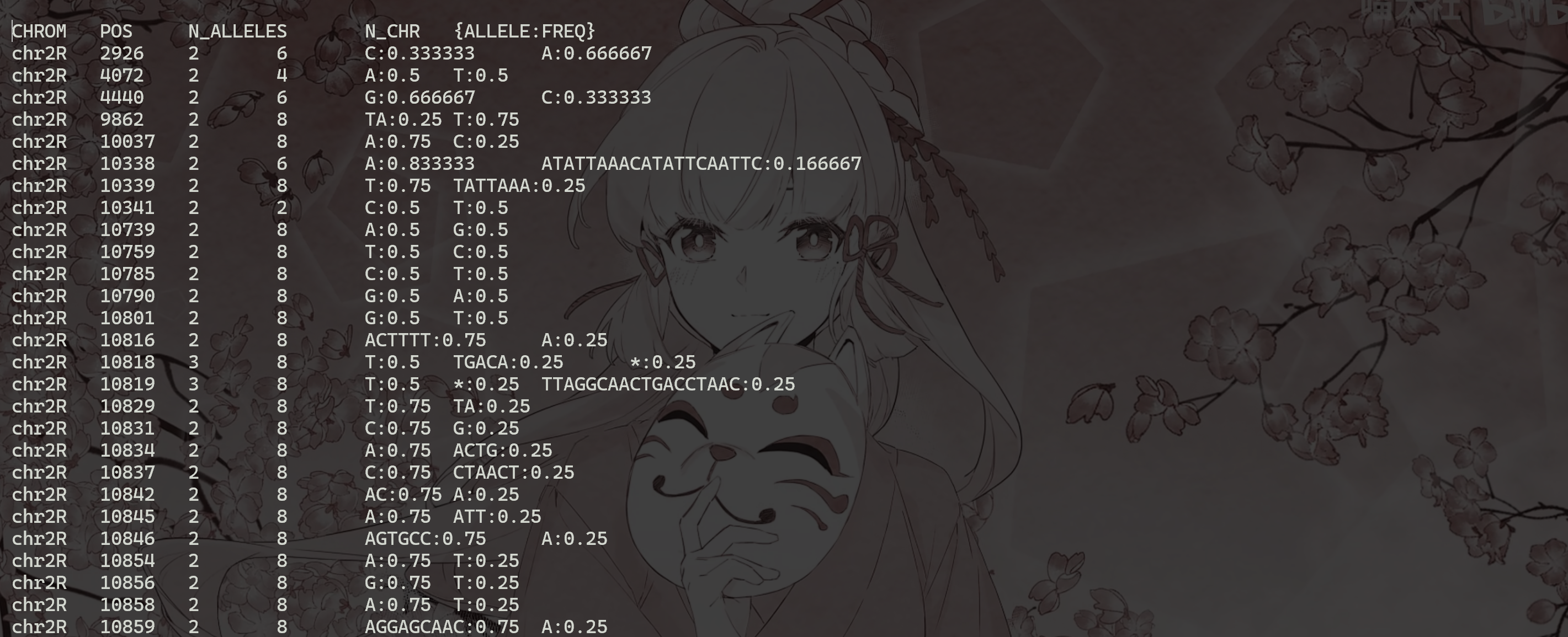
获取所有变体的等位基因频率并将其写入文件:
1 | |
比较两个VCF文件(将在文件fb.all.compare中打印出各种比较信息 ):
1 | |
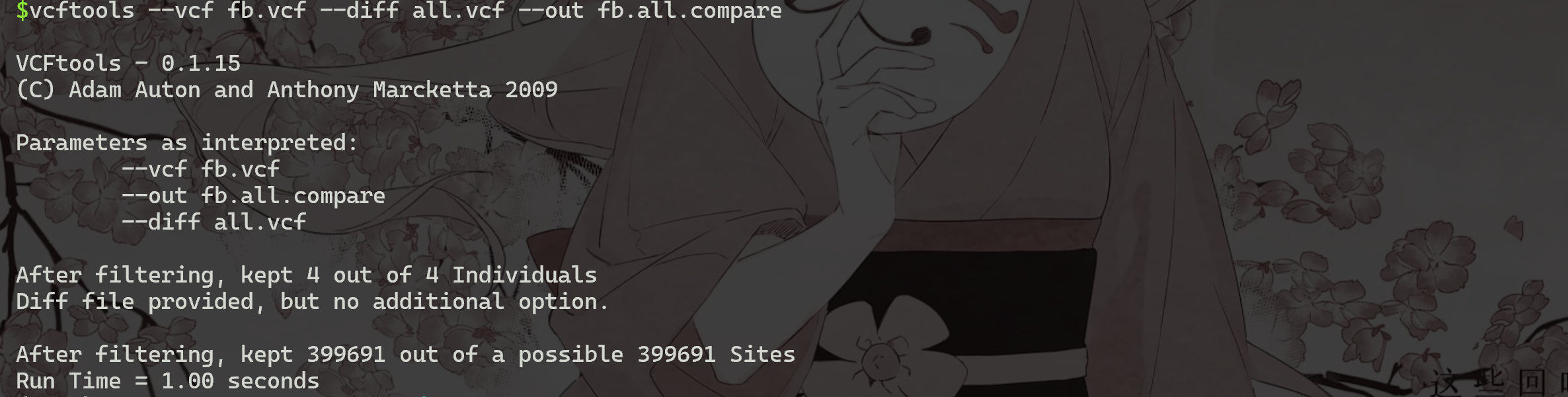
发现fb.vcf和all.vcf,都至少含有399691个SNP。之前实验可得GATK4发现了423493个SNP。
4. 实验总结
4.1 实验结论
- 在本次实验中,GATK4发现了423493个SNP,而FreeBayes发现399691个SNP。
- 使用GATK4,以all.vcf为原假设,在InDel中,大部分都是假阳性,只有8个是真阳性。 而使用FreeBayes,以fb.vcf为原假设,其中有79.2%的SNP都是真阳性,少部分为假阳性。
4.2 实验收获
- 使用samtools命令查看获取到的文件的比对状态汇总。
- 使用GATK4以及FreeBayes调用SNP体 。
- 学习利用IGV可视化BAM等文件,分析其生物学意义。
- 了解BAM与VCF等格式文件。