From Fastq data files to Read Count Matrix
1. 实验目的
练习从Fastq数据文件到Read Count矩阵的处理步骤:copy本实验的相关文件至工作目录,将Fastq文件进行质量控制和过滤,使用常见的质量控制工具FastQC来进行质量评估;使用适当的比对工具(如STAR)将Fastq文件的序列比对到参考基因组上,为每个序列分配基因信息;利用samtools软件对BAM文件建立索引,使用IGV软件来可视化BAM文件,并熟悉IGV软件操作与数据意义;编写bash脚本runSTAR.sh,根据比对结果计算每个基因的Read_Count;最后利用R对Gene_Count文件使用DESeq2绘制PCA图,展示基因表达数据的样本间差异。
了解perl_fork_univ.pl脚本运行STAR的多个作业的操作。
2. 实验准备
2.1 实验平台
Linux JSvr01 3.10.0-1160.80.1.el7.x86_64 #1 SMP Tue Nov 8 15:48:59 UTC 2022 x86_64 x86_64 x86_64 GNU/Linux
2.2 数据简述
| 数据 | 数据说明 | 数据来源 |
|---|---|---|
| R64.fa | Reference genome sequence file, in fasta format. | /work/cb-data/RNAseq/exercise1 |
| R64.gtf | Genome annotation file, in gtf format. | /work/cb-data/RNAseq/exercise1 |
| ERR458493.fastq.gz | RNA-seq data file, wt_sample1 | /work/cb-data/RNAseq/exercise1 |
| ERR458494.fastq.gz | RNA-seq data file, wt_sample2 | /work/cb-data/RNAseq/exercise1 |
| ERR458495.fastq.gz | RNA-seq data file, wt_sample3 | /work/cb-data/RNAseq/exercise1 |
| ERR458500.fastq.gz | RNA-seq data file, mu_sample1 | /work/cb-data/RNAseq/exercise1 |
| ERR458501.fastq.gz | RNA-seq data file, mu_sample2 | /work/cb-data/RNAseq/exercise1 |
| ERR458502.fastq.gz | RNA-seq data file, mu_sample3 | /work/cb-data/RNAseq/exercise1 |
| sample.txt | sample type | /work/cb-data/RNAseq/exercise1 |
2.3 软件配置
| 软件 | 版本 | 来源 |
|---|---|---|
| R | R version 4.3.1 | https://posit.co/download/rstudio-desktop/ |
| RStudio | RStudio Version 2023.06.2+561 | https://posit.co/download/rstudio-desktop/ Rstudio依赖R |
| Windows PowerShell | 5.1.22621.1778 | Windows系统自带 |
| FileZilla | 3.65.0 | https://filezilla-project.org/ |
| IGV | 2.6.3 | https://data.broadinstitute.org/igv/projects/downloads/2.6/ |
| notepad++ | 8.5.7 | http://notepad-plus-plus.org/ |
3. From Fastq data files to Read Count Matrix
本实验将进行RNA-Seq差异分析之前的数步操作:
首先将Fastq文件进行质量控制和过滤,使用常见的质量控制工具FastQC来进行质量评估;
然后使用STAR工具将Fastq文件的序列比对到参考基因组上,生成BAM格式的比对结果文件
其次使用samtools软件对BAM文件进行索引,使用IGV软件可视化BAM文件;
再次为每个序列分配基因信息:使用比对的结果文件(BAM文件)以及相应的基因注释文件,编写一个Bash脚本,其中包含运行比对工具(如STAR)以及计算每个基因的Read Count的步骤;
最后使用DESeq2软件包和R编程语言,对基因计数文件进行进一步的分析。可以使用DESeq2来绘制PCA图,以展示基因表达数据在样本间的差异。
3.1 Prepare the working directory
copy实验所需的数据至本人目录下,并创建工作空间。
1 | |
3.2 Examine the quality of the fastq data files
QC过程,对数据进行质量评估
(1)对fastq文件使用命令fastqc
使用质量控制工具FastQC来进行质量评估
1 | |
(2)使用Filezilla下载生成的ERR458493_fastqc.html,查看分析结果

1 | |
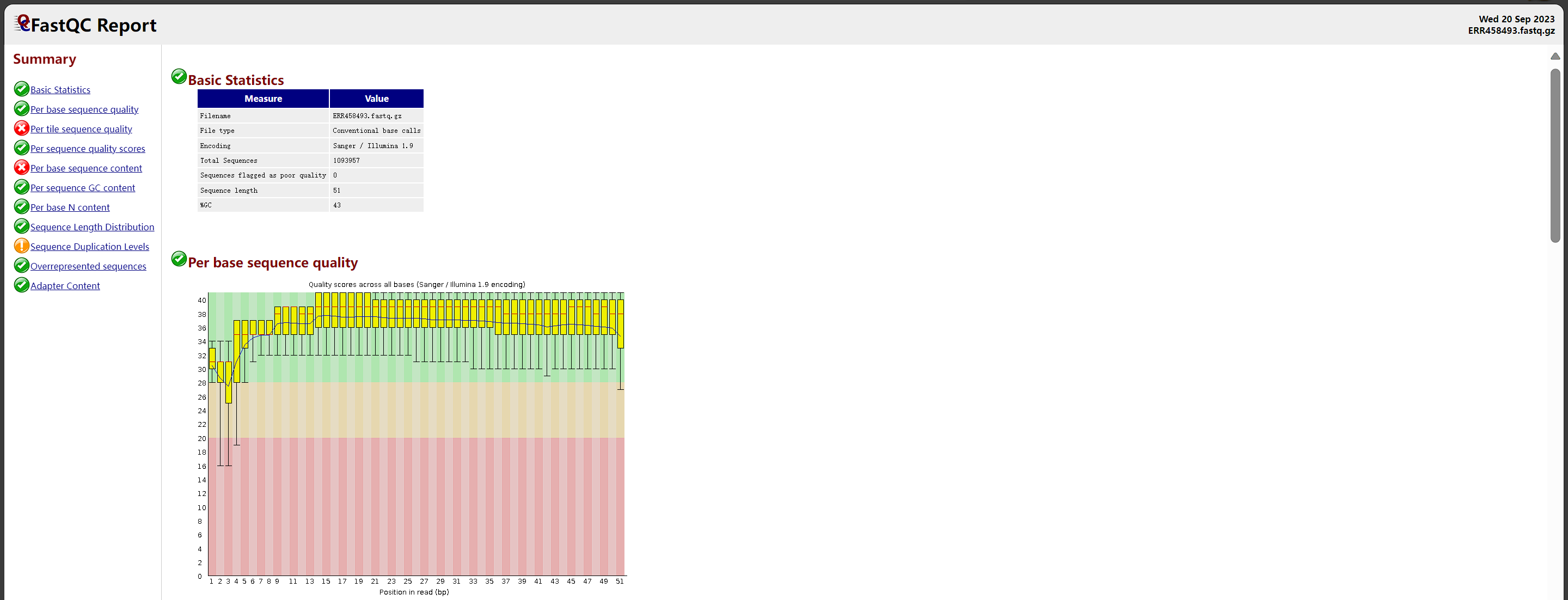
由Per base sequence quality分析可知,整体质量不错,得分总体处于28以上,当序列长度为2,3,4bp时,误差较大,建议舍去。
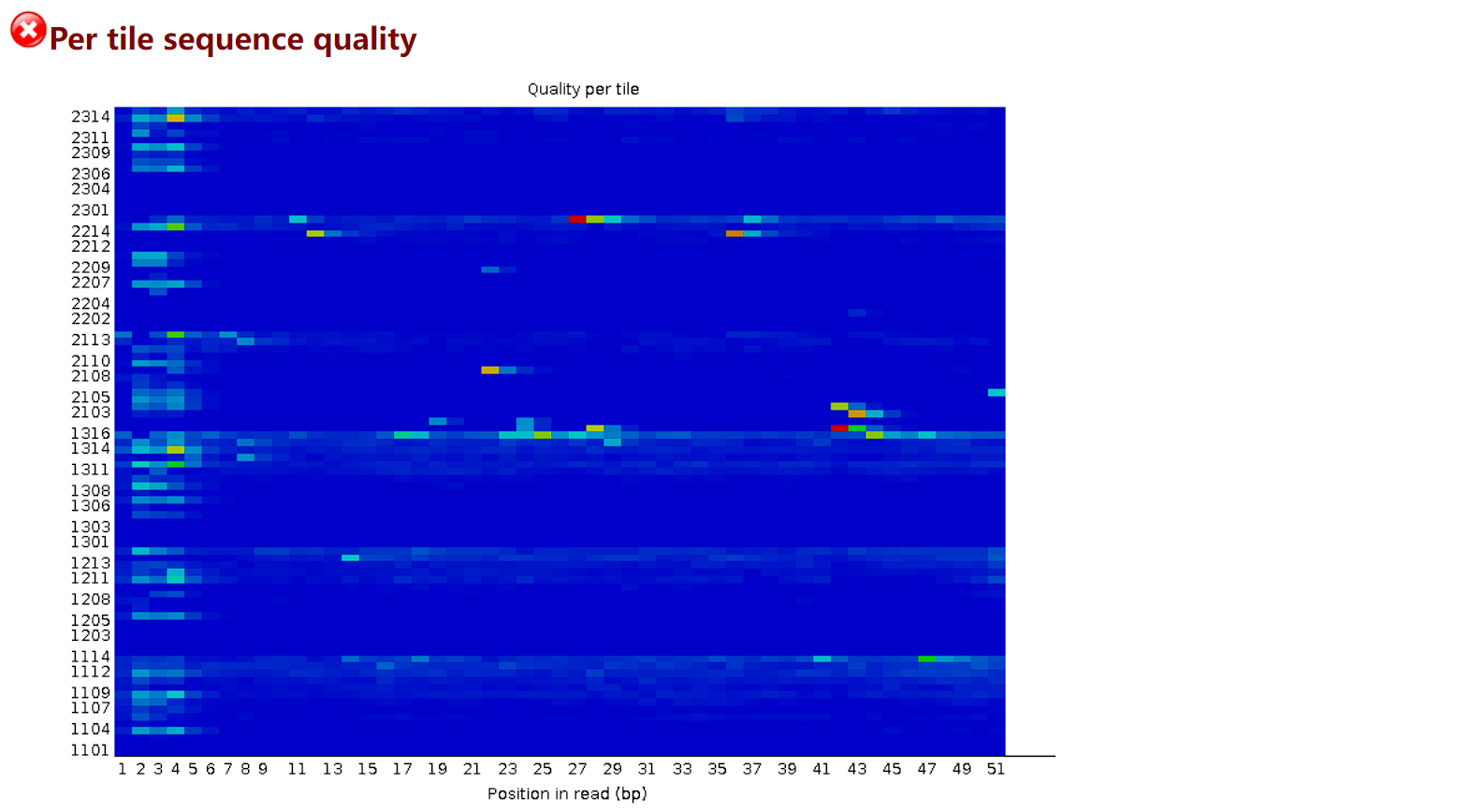
对于每个tile的测序质量,用热图进行展示,蓝色表明该tile的测序质量好,红色表明该tile的测序质量差。
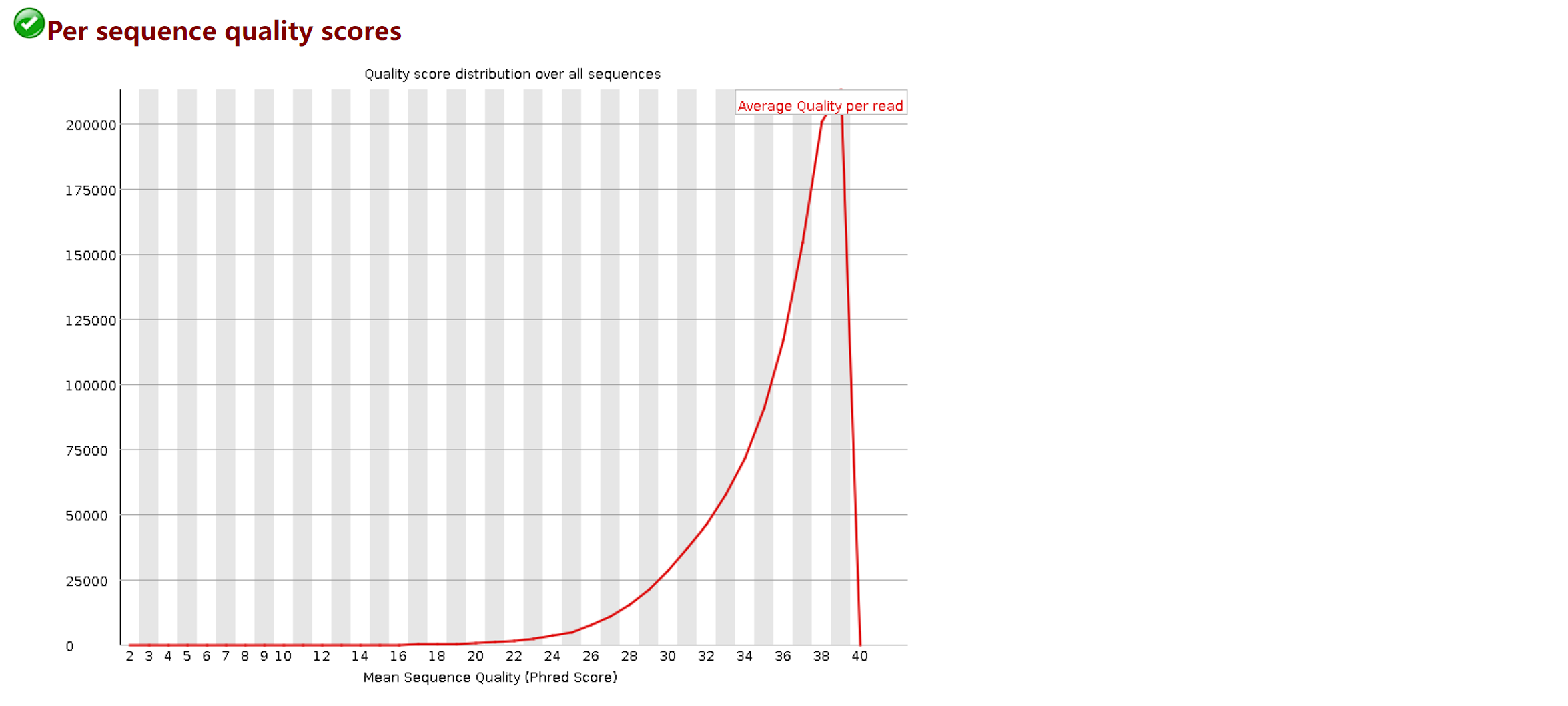
横坐标为reads的平均测序质量,纵坐标为序列数,峰值在39处,测序质量好
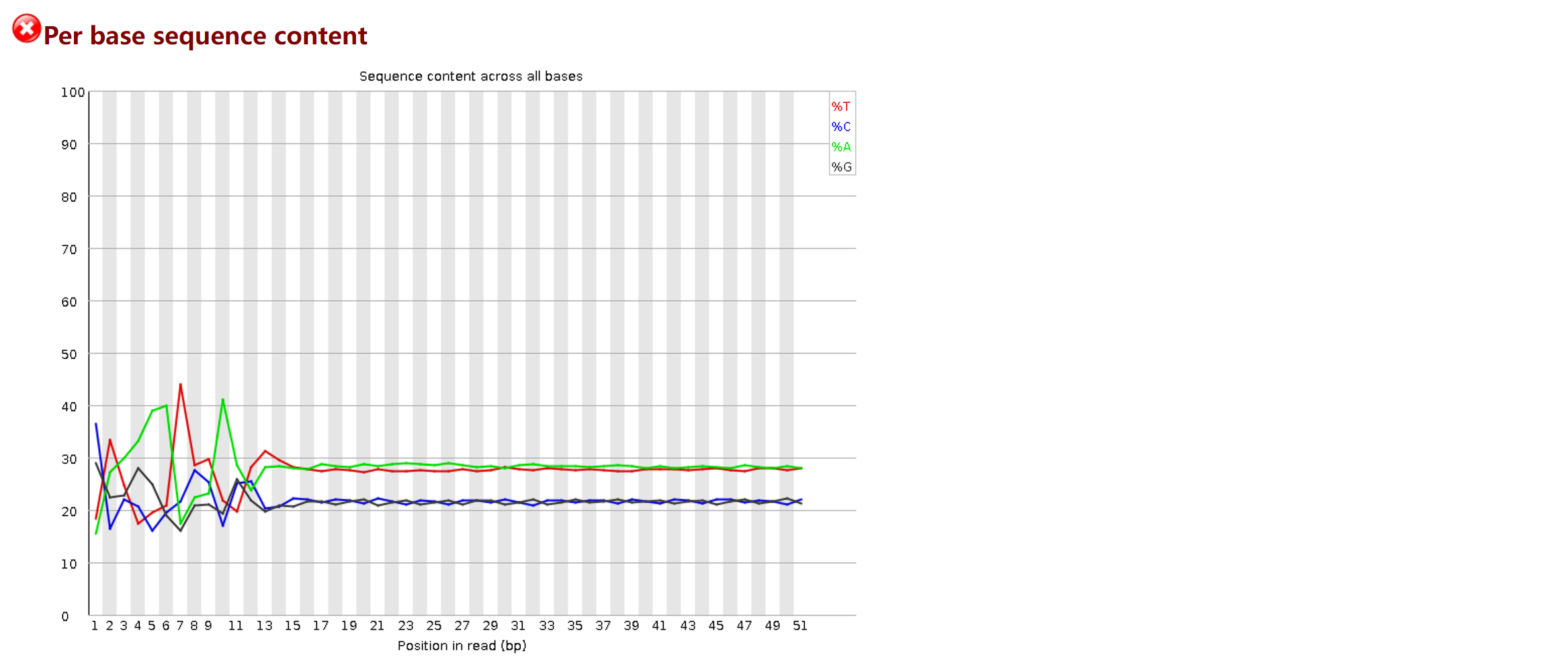
横坐标为序列长度,纵坐标为各碱基的百分比,可以看出序列长度较短时,碱基分布的偏倚,文库的构建过程存在问题。
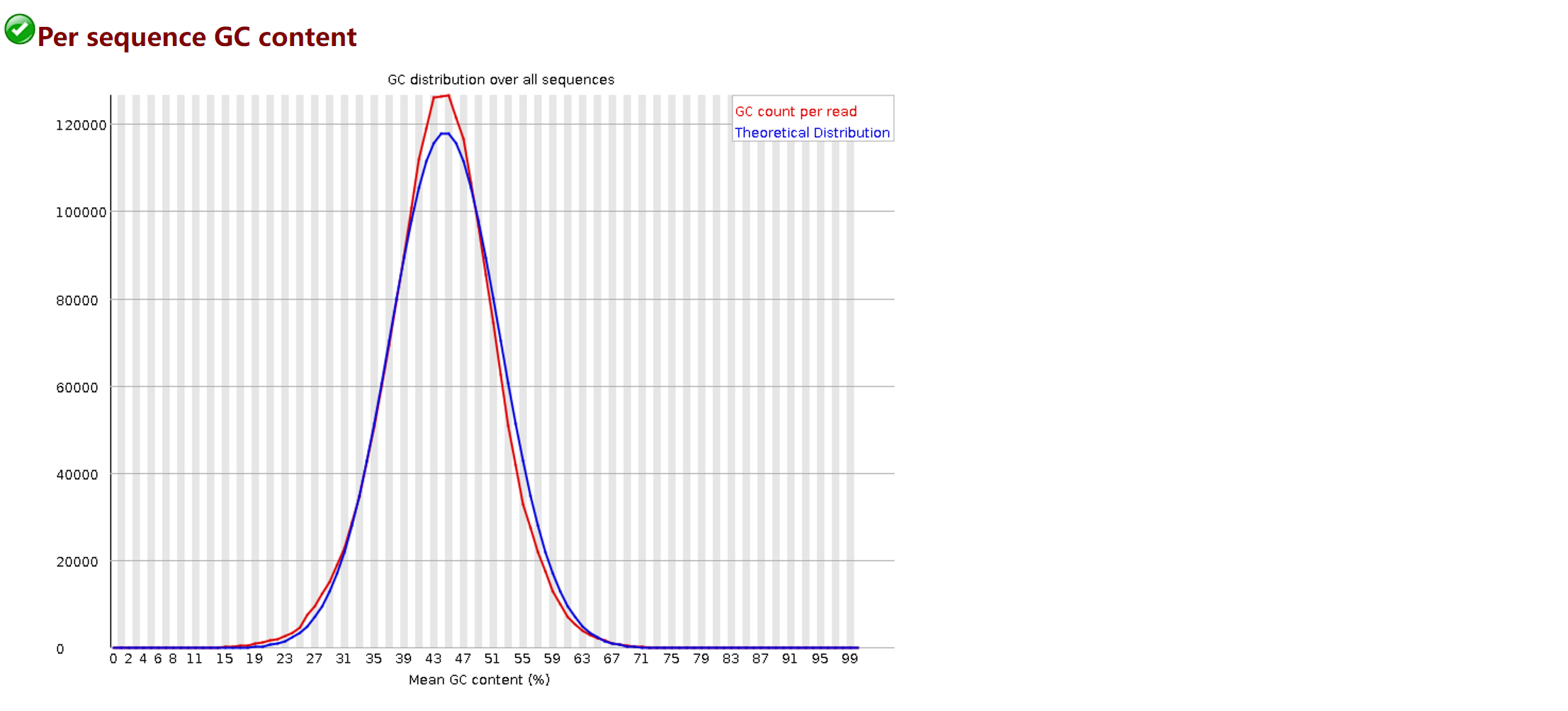
横坐标为GC含量,纵坐标为序列,数图中红色的线是实际的GC含量分布,蓝色的线是理论上的正态分布曲线,可知CG含量总体符合正态分布。
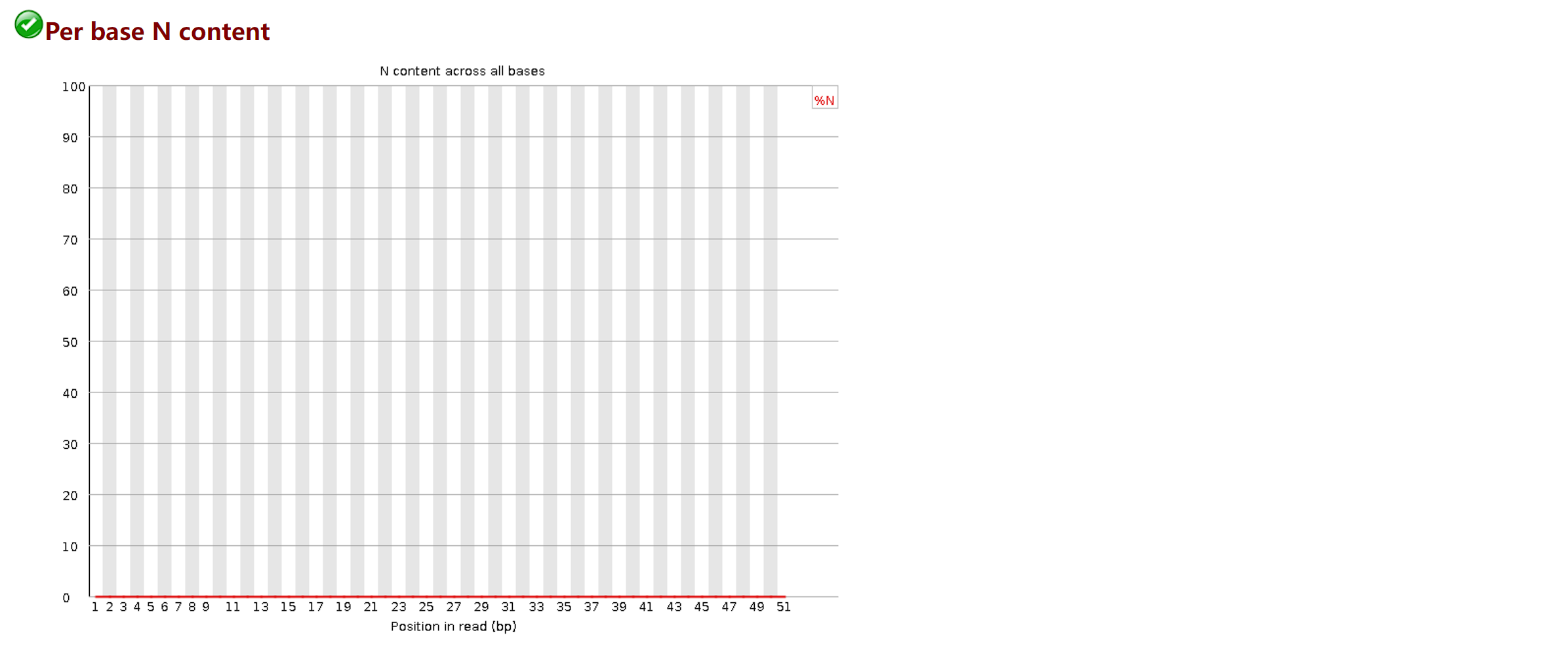
这部分内容给出N碱基的比例分布图,横坐标为序列长度,纵坐标为N碱基的比例,当测序仪无法识别具体是哪种碱基时,就会给出N, N比例越小越好。该图质量较好。
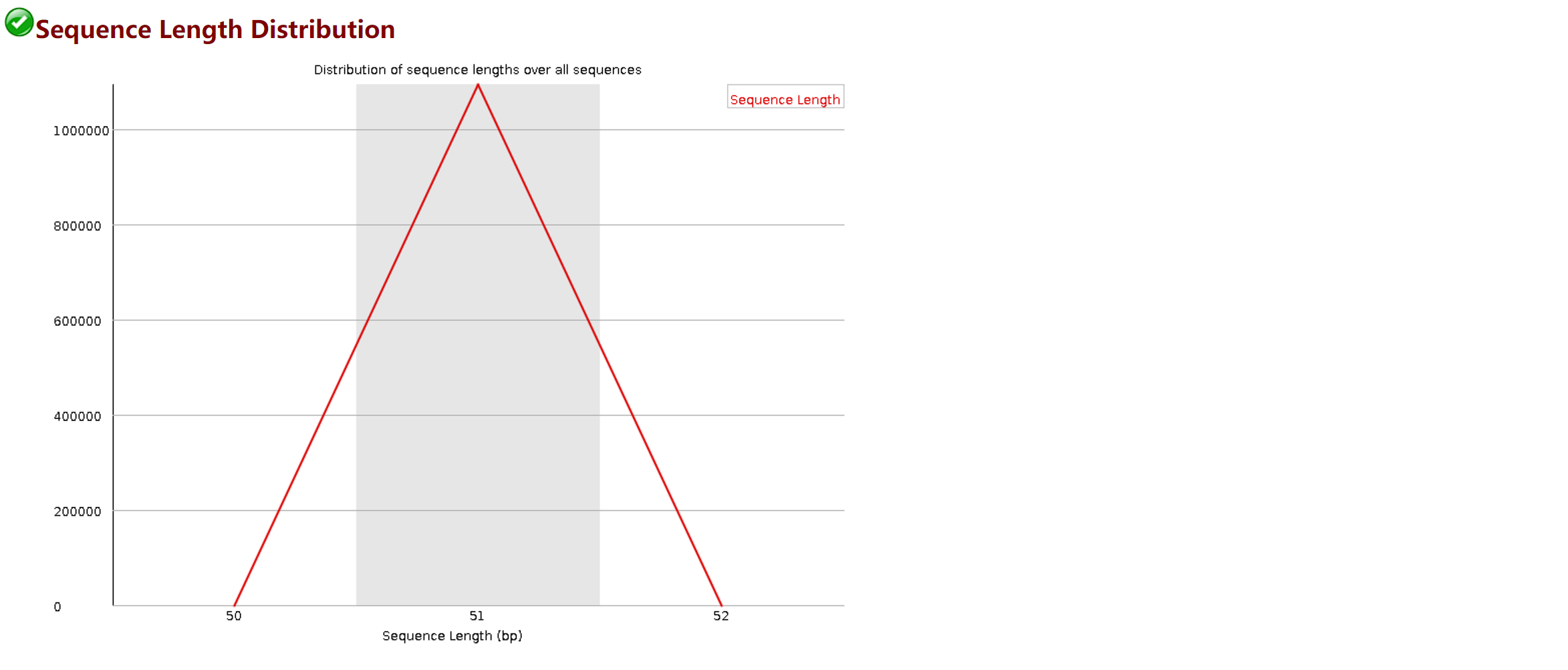
这部分内容给出序列的长度分布,横坐标为序列长度,纵坐标为序列条数,峰值在51bp处。
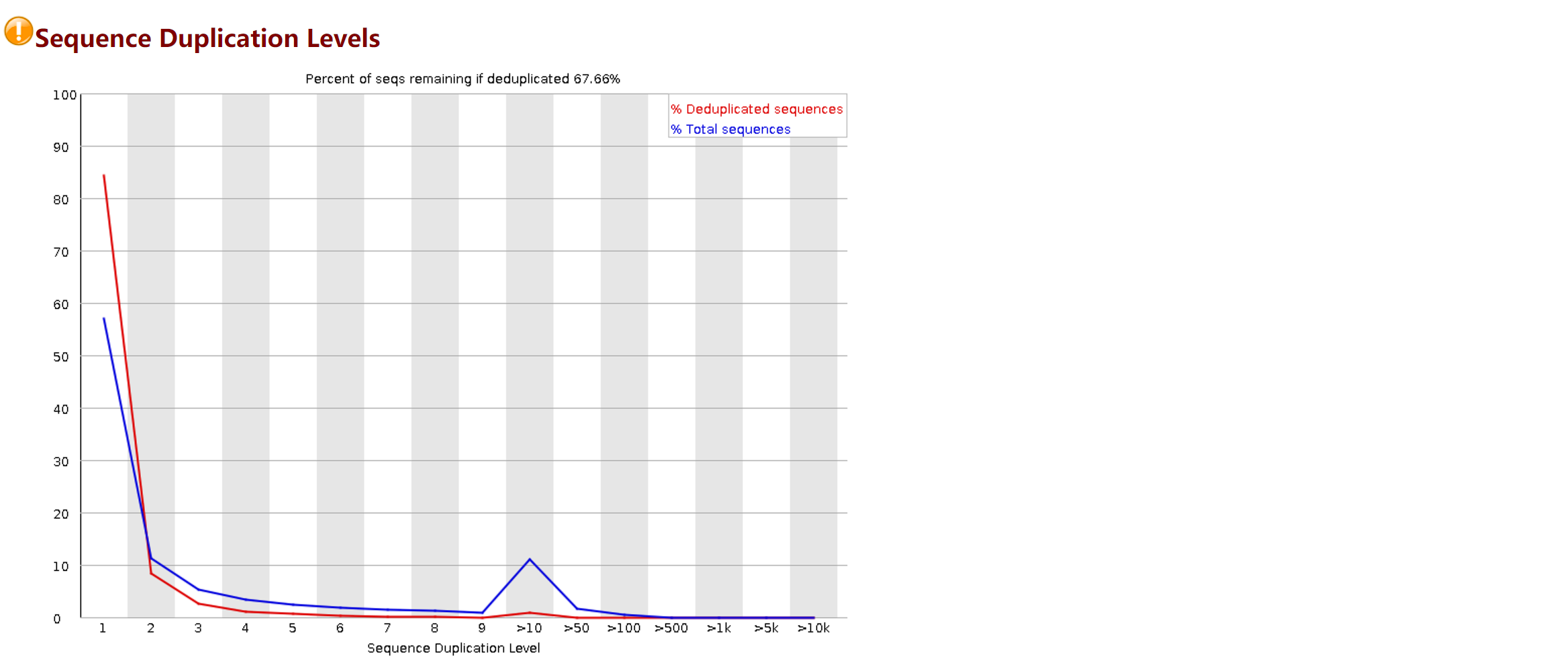
这部分给出重复序列分布图,横坐标为重复的次数,纵坐标为序列所占百分比。基因组覆盖度越高,测序得到的序列重复比例会越低;在文库构建过程中,如果某些片段PCR扩增的比例大于随机扩增的比例,会导致重复序列比例高。
这部分内容给出序列中包含的adapter 序列的情况。
3.2 Run read mapping software
使用STAR工具将Fastq文件的序列比对到参考基因组上
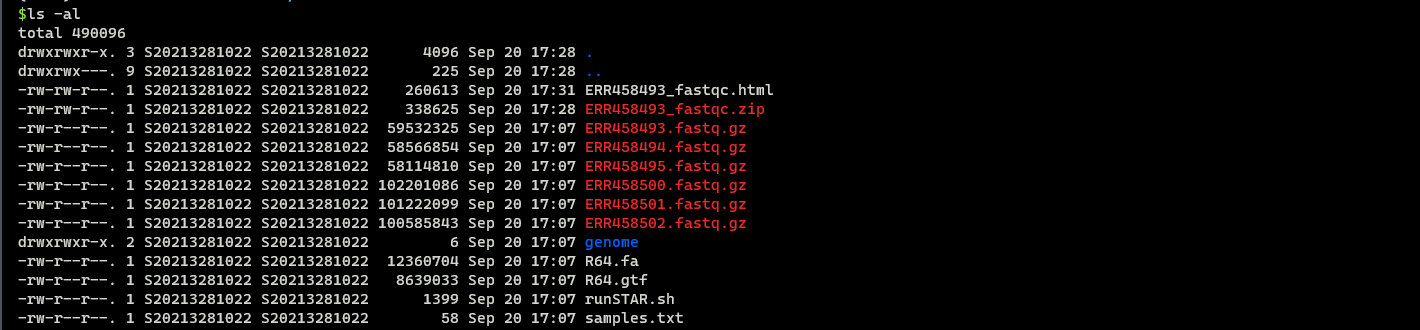
(1)检查文件及文件基本信息
1 | |
1 | |
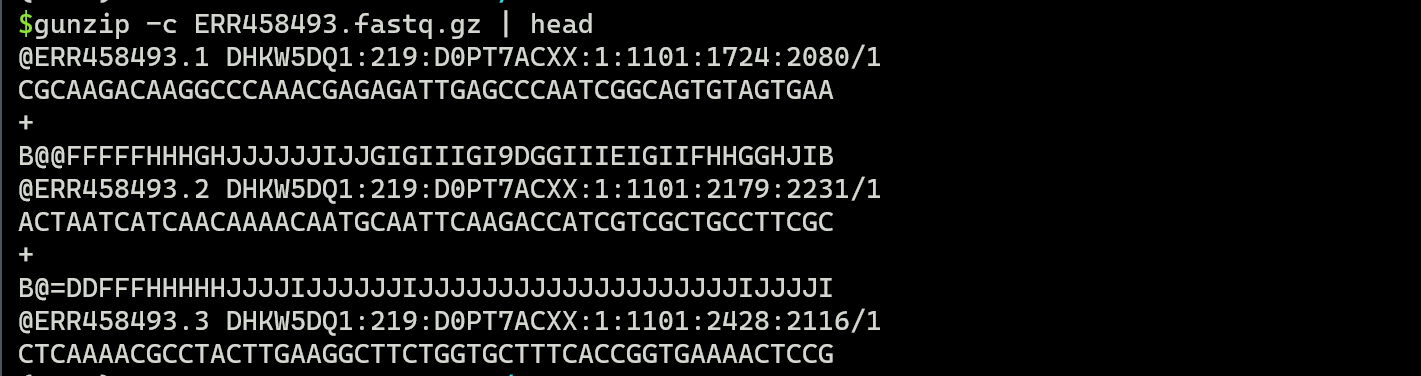

由于每个序列读取在 Fastq 文件中占用 4 行,因此行号除以 4 给出了文件中的排序读取数。reads数为:4375828/4=1093957

展现fa格式文件,共202645行,仅展现前5行

展现gtf格式文件共41884行,仅展现前5行,以下为各列含义:
- seq_id:序列的编号,一般为chr或者scanfold编号,每条染色体拥有一个唯一的ID。
- source: 注释的来源,代表基因结构的来源,可以是数据库的名称,比如来自
RefSeq数据库,也可以是软件的名称,比如用GeneScan软件预测得到,当然,也可以为空,用.点号填充。- type: 代表区间对应的特征类型
- start:该基因或转录本在参考序列上的起始位置。
- end: 该基因或转录本在参考序列上的终止位置。
- score: 得分,软件提供了统计值,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值,“.”表示为空。
- strand: 代表正负链的信息,
+表示正链,-表示负链,?表示不清楚正负链的信息,当正负链信息没有意义时,可以用.填充。- phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为0、1、2
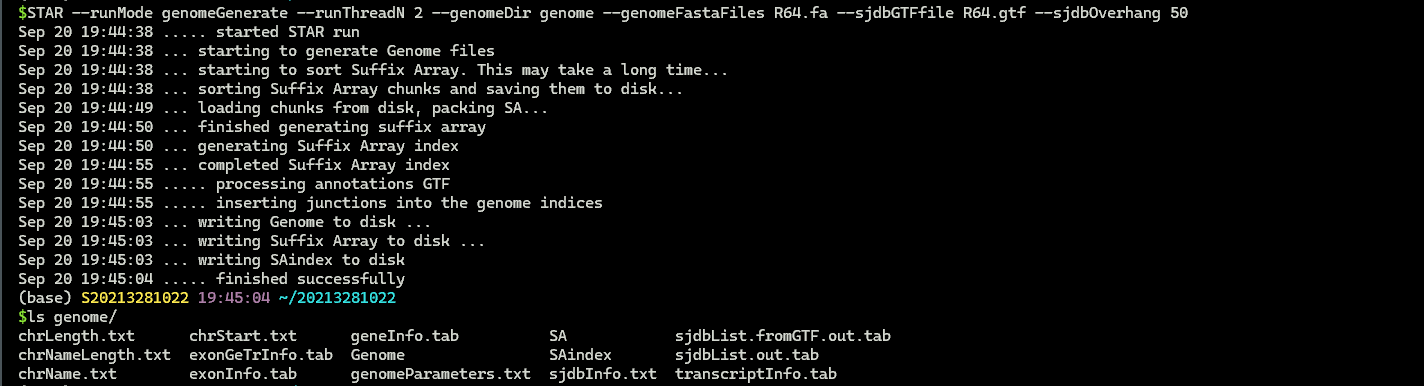
(2)使用STAR将读段映射到参考基因组
1 | |
1 | |
1 | |
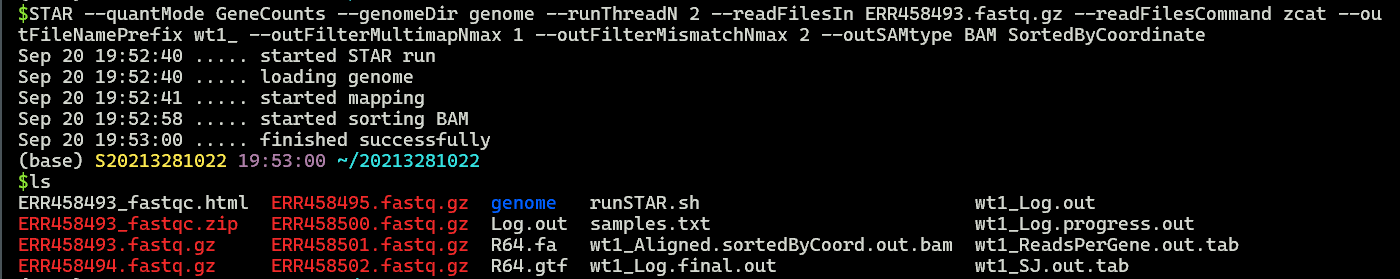
1 | |

共7131行,仅展现前5行
3.4Visualize the BAM file with IGV
利用samtools软件对BAM文件建立索引,使用IGV软件来可视化BAM文件
(1)为bam文件编制索引:我们将使用IGV软件来可视化bam文件。为了让IGV读取BAM文件,需要对“.BAM”文件进行索引。我们将使用samtools软件
1 | |
(2)使用FILEZILLA将“.bam”、“.bai”、“R64.fa”和“R64.gtf”文件下载到笔记本电脑。
(3)安装IGV_2.6.3
https://data.broadinstitute.org/igv/projects/downloads/2.6/
(4)创建自己的基因组
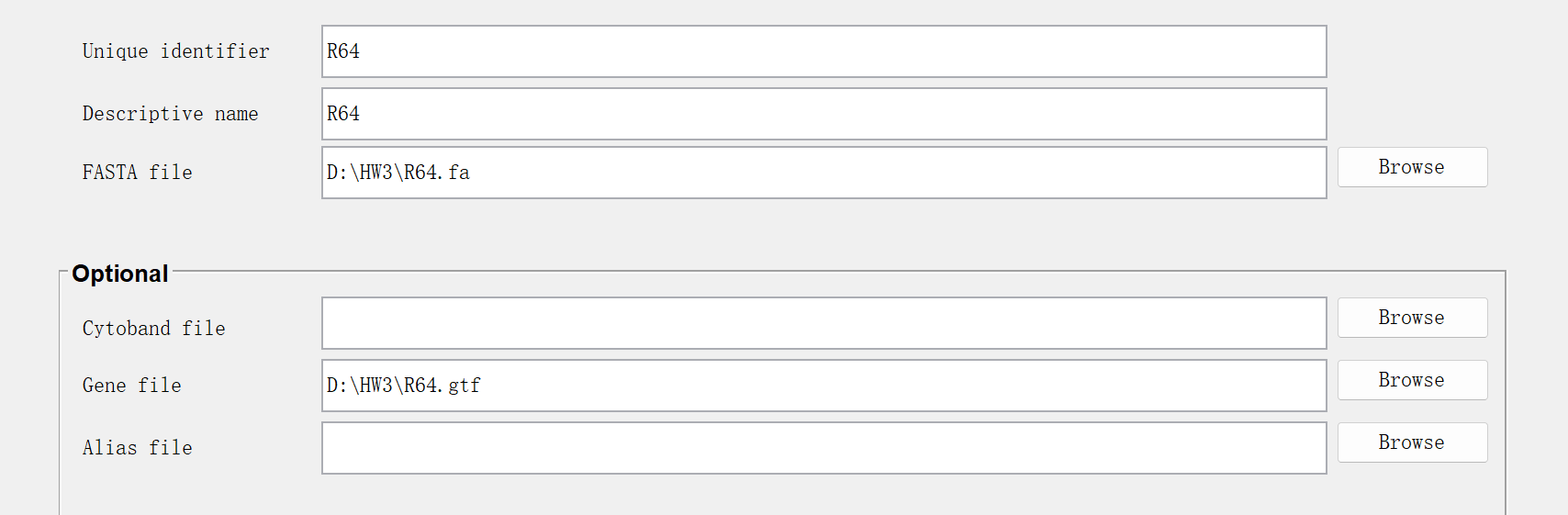
生成R64.genome文件
(5)导入文件后,检测区域II:265,593-282,726
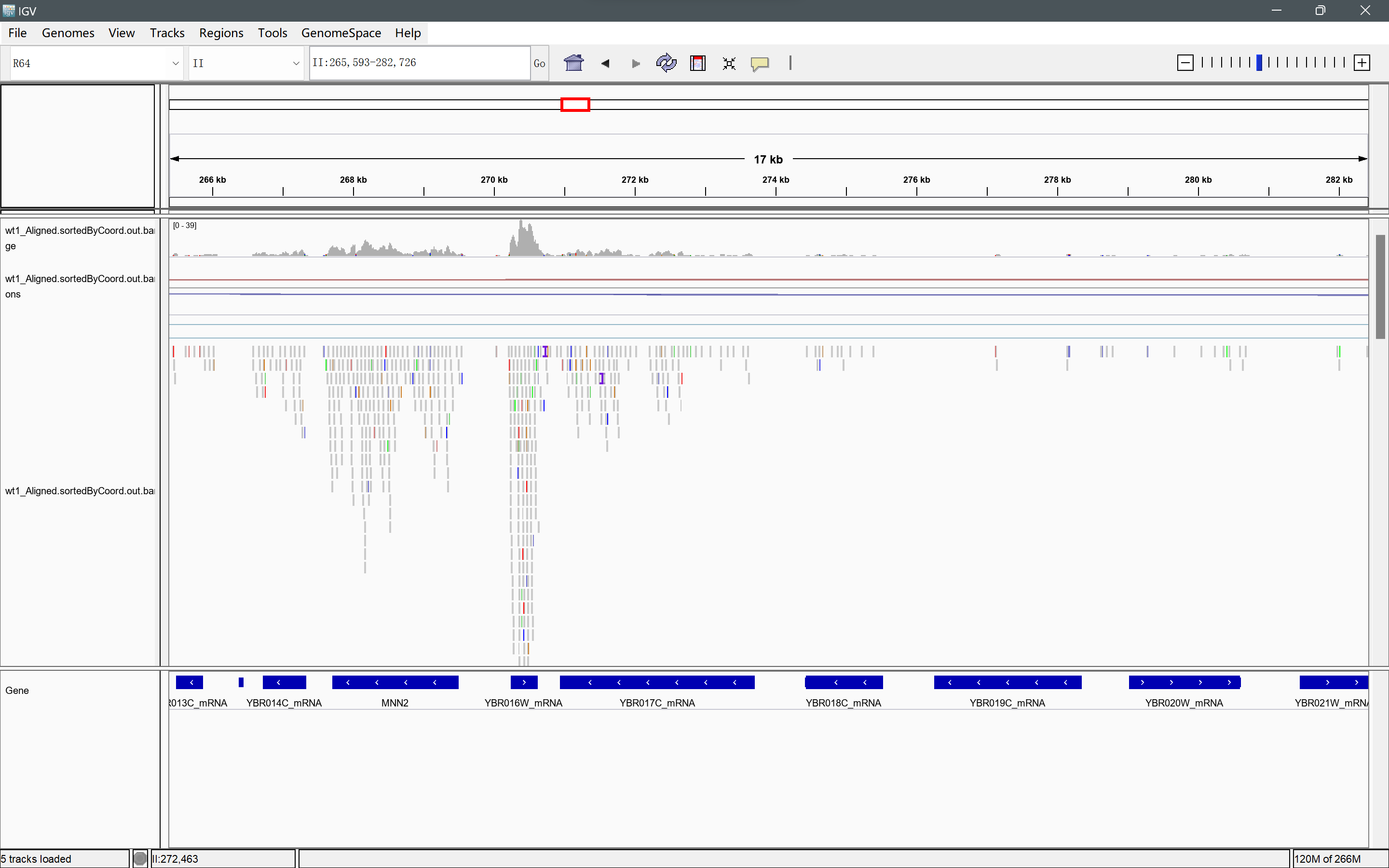
展现了II号染色体上的265,593至282,726区域的基因与reads数。峰值位于268kb与270.5kb处,可知YBR016W_mRMA与MNN2表达量较其他基因高,同时考虑外显子长度,YBR016W_mRMA较短故可认为表达量极高,而YBR020W_mRMA, YBR019C_mRMA, YBR018C_mRMA,YBR017C_mRMA长度长但reads数很少,可认为表达水平较低。同时reads除了match部分,还有较多的SNP位点、插入与缺失位点,大多处于reads的边缘。处于正链的基因箭头向右,处于负链的基因箭头向左。
3.5 Run the commands in a shell script
在典型的RNA-seq实验中,有许多样品,可能需要数小时才能完成比对。可以做两件事来加快计算速度: 创建一个批处理命令(shell script)来处理所有文件; 使用 STAR 的“共享内存”功能。
(1)下载文本编辑器
http://notepad-plus-plus.org/
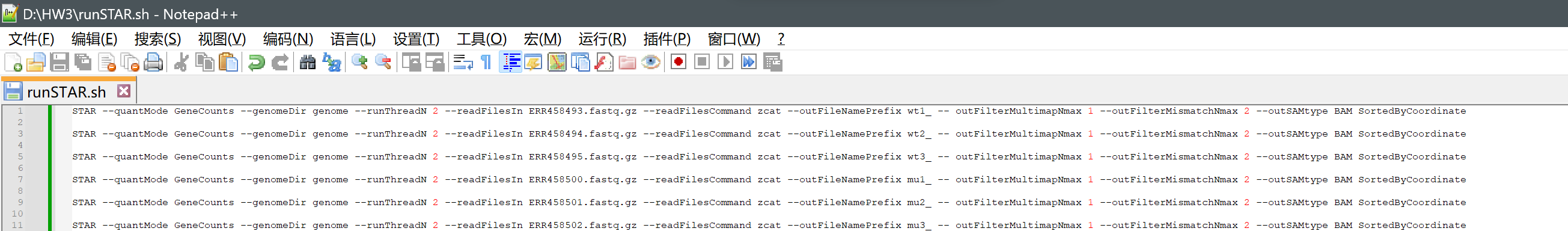
(2)创建脚本runSTAR.sh
1 | |
–quantMode GeneCounts: 这个参数告诉STAR使用基因计数模式进行分析,即生成基因计数信息。
–genomeDir genome: 这个参数指定基因组目录的路径,STAR将在该目录中查找索引文件和其他相关文件。
–runThreadN 2: 这个参数指定并行处理的线程数。在这种情况下,STAR将使用2个线程进行处理。
–readFilesIn: 这个参数指定输入的FASTQ文件的路径。
–readFilesCommand zcat`: 这个参数告诉STAR使用zcat命令解压缩输入的FASTQ文件。在这个例子中,它使用zcat命令来解压缩。
–outFileNamePrefix : 这个参数指定输出文件的前缀。
–outFilterMultimapNmax 1: 这个参数指定最大允许的多映射数。在这个例子中,允许的最大多映射数为1。
–outFilterMismatchNmax 2: 这个参数指定最大允许的错配数。在这个例子中,允许的最大错配数为2。
–outSAMtype BAM SortedByCoordinate: 这个参数指定输出的SAM/BAM文件类型以及按坐标排序。在这个例子中,输出将是按坐标排序的BAM文件。
通过运行这个STAR命令,您将得到基因计数信息和按坐标排序的BAM文件,这对于后续的基因表达分析非常有用。请确保您已经正确安装和配置了STAR软件,并且已经生成了相应的基因组索引。
然后利用FileZilla拷贝文件至服务器
(3)启用screen,运行脚本
1 | |
1 | |
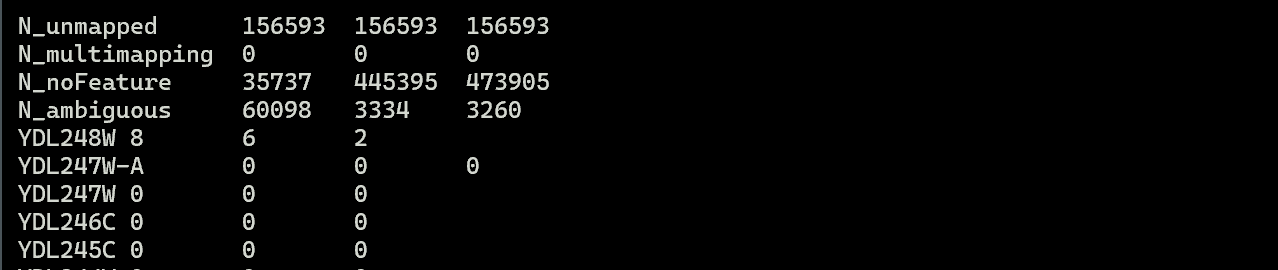
(4)每个样本一个文件(*_ReadsPerGene.out.tab)。将这6个文件合并为一个文件进行统计分析。
1 | |
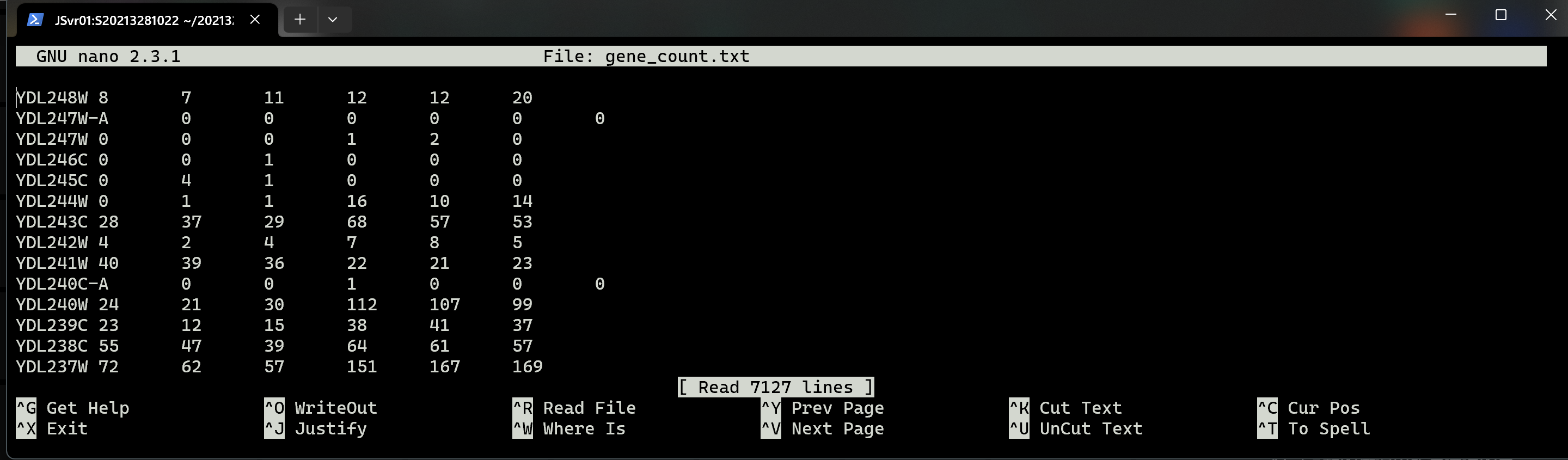
共7127行。
3.6 Load the matrix into R and make PCA Plot with DESeq2
PCA(Principal Component Analysis)是一种常用的多变量数据分析方法,可以帮助我们理解和解释生物学数据的特征和变异。PCA可以从数据中找出主要的变异模式,并将原始数据映射到新的低维空间中,使得数据的变异尽可能地集中在少数几个主成分上。
(1)练习打开sample文件
1 | |

(2)用Filezilla传输gene_count.txt和samples.txt文件,在RStudio上导入文件,使用DESeq2绘制PCA图
1 | |
我们使用散点图来表示PCA的结果。选择前两个主成分,第一主成分具有最大的方差,可以看到PC1将mu与wt分得很开,具有良好的区分效果,而PC2则难以将其区分。类内较为紧密,类间间距大,数据质量较好。
3.7 Use the “shared memory” feature load of STAR
STAR提供了一个功能,允许将基因组数据库预加载到共享内存空间中,所有STAR比对作业都可以使用该空间。
(1)将基因组加载到数据库中并保存
1 | |

(2)创建脚本
像在步骤3.5中那样,使用STAR比对命令制作一个shell脚本。将这两个参数添加到每个STAR命令中:“–genomeLoad LoadAndKeep–limitBAMsortRAM 400000000”。genomeLoad指示STAR使用共享内存,limitBAMsortRAM指示STAR限制4GB用于bam排序步骤。
1 | |
1 | |
(3)完成后,确保从共享内存中删除基因组数据库。否则它会留在那里。
1 | |

4. 实验总结
4.1 实验结论
(1)使用FastQC对Fastq文件来进行质量评估,由Per base sequence quality分析可知,整体质量不错,得分总体处于28以上,当序列长度为2,3,4bp时,误差较大,建议舍去。
(2)使用STAR将测序读数与索引基因组比对输出排序的 bam 文件
(3)使用IGV软件来可视化bam文件,需要对“.BAM”文件进行索引。可由IGV可视化BAM文件可知知YBR016W_mRMA与MNN2表达量较其他基因高,同时考虑外显子长度,YBR016W_mRMA较短故可认为表达量极高,而YBR020W_mRMA, YBR019C_mRMA, YBR018C_mRMA,YBR017C_mRMA长度长但reads数很少,可认为表达水平较低。
(4)利用R对Gene_Count文件使用DESeq2绘制PCA图,展示基因表达数据的样本间差异。可以看到PC1将mu与wt分得很开,具有良好的区分效果。类内较为紧密,类间间距大,数据质量较好。
4.2 实验收获
注意到数据分析前的QC过程。学会使用STAR将Fastq文件的序列比对到参考基因组上,为每个序列分配基因信息。学会利用samtools软件对BAM文件建立索引。学会使用IGV软件来可视化BAM文件,并熟悉IGV软件操作与数据意义。了解PCA图的绘制流程与其中的生物学意义。